US Lab in Kenema Sierra Leone; Ft Detrick and Metabiota Possible Roles. 2023-07-14
Main Man

by Sam Husseini and Jonathan Latham, PhD
Between 2014 and 2016, West Africa endured an Ebola epidemic that was easily the largest and deadliest in history. Over 29,000 people were infected and more than 11,000 died in what was also an economic and social calamity.
The countries most afflicted were Sierra Leone, Liberia, and Guinea; but lives were also lost far afield. Ebola cases were detected in Nigeria, Senegal, Mali, Spain, the UK, and the U.S. This international spread unleashed its own, albeit fairly short-lived, panic.
The infectious agent that caused the outbreak was a filovirus, the Zaire species of Ebola virus (sometimes called ZEBOV and sometimes just Ebola), which has a fatality rate of up to 90 percent (Feldmann and Geisbert, 2011).
The orthodox story of the outbreaks’ origin remains the one given at the time by the global media. In the U.S., the Atlantic ran “The Beautiful Tree, the Bats, and the Boy Who Brought Ebola.” The UK Independent led with “Ebola outbreak: Two-year-old boy from tiny Guinea village was first to be infected after playing with bats in tree stump.” The BBC filed: “First Ebola boy likely infected by playing in bat tree” and Canada’s Toronto Star informed its readers “Scientists trace Ebola outbreak to a tree where children play“. While disagreeing whether it was a “stump” or a “beautiful tree”, the media all concurred on one African boy and the bats.
The primary source for these accounts was a research paper that appeared on December 30th, 2014 in the journal EMBO Molecular Medicine (Saéz et al., 2014). Titled “Investigating the zoonotic origin of the West African Ebola epidemic“, the paper begins:
“The severe Ebola virus disease epidemic occurring in West Africa stems from a single zoonotic transmission event to a 2‐year‐old boy in Meliandou, Guinea.”
Their claimed patient zero, Emile Ouamouno, allegedly caught Ebola after playing with, or maybe hunting, bats.
Perhaps dissuaded by the definitive opening sentence, the media seems not to have enquired at the time into what evidence supported this narrative. This was perhaps a mistake.
Granted, there seemed grounds for confidence in these scientists. The senior author was Fabian Leendertz of the prestigious Robert Koch Institute in Germany. Leendertz is a renowned virus hunter best known as a member of the WHO team that reported, in March 2021, on the origins of COVID-19.
Missing evidence: the search for a zoonosis in Meliandou
For their investigation, the Leendertz team surveyed bats in the area surrounding Meliandou. Bats were chosen since they were considered the presumptive reservoir host of Zaire Ebola viruses (Leroy et al., 2005; Leroy et al, 2009; Pigott et al., 2014).
Samples of bat blood and bat tissue were collected from 159 individuals of 13 bat species. However, the results were uniformly negative:
“No EBOV RNA was detected in any of the PCR-tested bat samples [and] attempts to demonstrate the presence of IgG antibodies against Ebola viruses were inconclusive (data not shown).”
Trying another tack, the authors knew that outbreaks of Zaire Ebola are sometimes correlated with mass die-offs of forest mammals (Walsh et al., 2003; Leroy et al., 2004). However, no evidence for mammal declines could be found near Meliandou:
“[these results] suggest they have not experienced a major decline; in fact, carnivore and chimpanzee (Pan troglodytes verus) populations may have increased.”
All-told, the Leendertz team found no sign whatever of Ebola in the area around Meliandou.
What of Emile? The Leendertz team concluded that Emile and his mother, who was eight months pregnant at the time, were both Ebola victims, as was a sister who also died. They did not note, however, that Emile’s primary caregiver at the time (probably because of the pregnancy) was his father, who never became sick.
Moreover, no blood or other samples were ever taken from any of these suspected cases. Hence no laboratory evidence exists for any of them having had Ebola and so these diagnoses rest on symptoms alone.
This is highly significant because the symptoms of Ebola are very variable. Even when severe, they overlap with many diseases that are common in West Africa, including malaria, cholera, and Lassa fever. It is therefore generally agreed that Zaire Ebola cannot be diagnosed without genome sequencing or other lab tests (Gire et al., 2014). These tests were not available until much later in the outbreak.
Thus the Leendertz investigation detected no Ebola in bats or other animals in the vicinity of Meliandou, nor did they uncover any other evidence that an Ebola outbreak had occurred in the region. They also found no evidence that Emile or his immediate contacts had Ebola infections; nor even was there a clear indication that Emile had any contact with a bat or the now infamous tree. Such findings did not warrant anything like the absolute certitude of the opening sentence of their report.
Rather, the evidence collected by the Leendertz team was, if anything, against Ebola having been in Meliandou at that time.
Independent evidence against Ebola in Meliandou
Chernoh Bah, an independent journalist from Sierra Leone, wrote a book on the 2014 Ebola outbreak and visited Meliandou. Bah found that:
“Local health workers still think malaria may have been the actual cause of his [Emile’s] death.”
While in Meliandou, Chernoh Bah also interviewed Emile’s father. According to Bah, the Leendertz team (who never claimed to have interviewed the father) made a crucial error:
“The child was actually 18 months old when he died.”
Bah confirmed this assessment in an interview with Independent Science News. This age is also accepted by the US CDC and was independently confirmed by Reuters journalists, who also interviewed Emile’s father. The age question, it should be noted, is crucial to the entire outbreak narrative. As Emile’s father told Reuters:
“Emile was too young to eat bats, and he was too small to be playing in the bush all on his own. He was always with his mother.”
Bah also identified another apparent error: that Emile had four siblings who never became sick. These siblings are not mentioned anywhere in the scientific literature.
The wider context is also important for weighing these contending accounts. Some previous outbreaks of Zaire Ebola have been linked with hunting, but only once has an Ebola outbreak been linked to handling or consumption of bats, and then only tentatively (Leroy et al., 2009). Thus, although catching and eating bats is common in parts of Africa, a clear precedent for their passing Ebola to humans does not exist.
Further, although some bats appear to carry antibodies against Ebola viruses, only intact Bombali Ebola (a different virus species in the Ebola genus) has ever been isolated from a bat, despite intensive searches (Leroy et al., 2005; Pigott et al., 2014; Goldstein et al., 2018; Forbes et al., 2019). Bombali is a species of Ebola that does not infect humans.
Taken together, this suggests that bats rarely carry Ebola viruses and when they do it is in small quantities. This context makes it somewhat surprising that Saéz et al. ascribed the 2014 outbreak (without supporting evidence) to contact with bats. Indeed, Fabian Leendertz now doubts that bats are true reservoirs of Ebola viruses (Leendertz et al., 2016).
Given the general want of evidence, one wonders by what exact process such poorly supported claims were transmuted into international headlines.
The missing epidemiology connecting Emile to the first proven Ebola cases
Emile Ouamouno died in Meliandou, Guinea, on Dec 6th, 2013 (Saéz et al. 2014; Baize et al., 2014).
Relying on hospital records and interviews, a putative transmission chain connecting Emile to (most of) the earliest confirmed cases was published in the New England Journal of Medicine (Baize et al., 2014).
The proposed epidemiological chain is shown in full in figure 2 (reproduced from Baize et al., 2014). In figure 2, suspected cases are indicated by an ‘S’ while patients confirmed by laboratory test are denoted with a ‘C’ for ‘confirmed’ (and also given a red spot). This makes Emile ‘S1’ in the standard narrative.
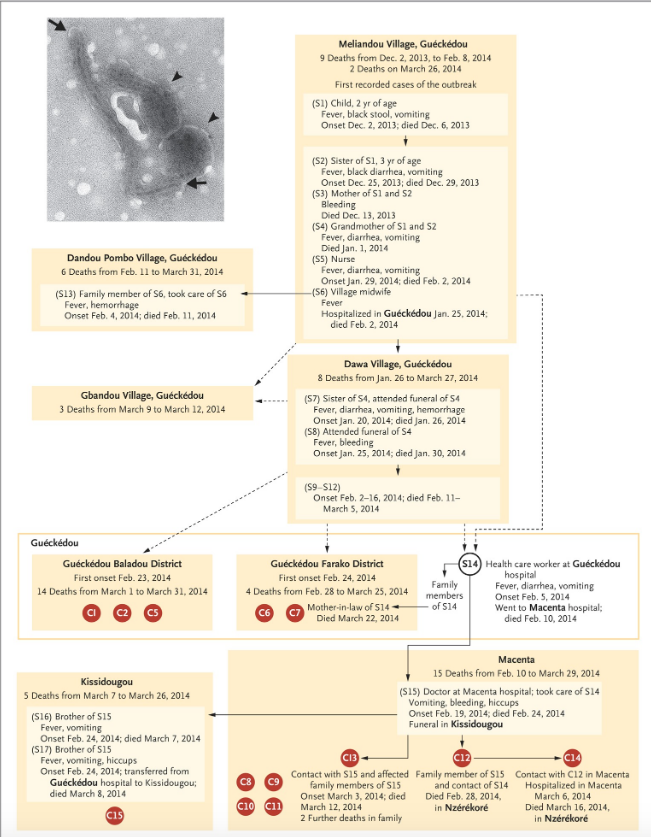
These authors acknowledge that the dashed connections in figure 2 represent epidemiological links that “are not well established”. It is thus important to note that Emile connects to the first confirmed cases only via dashed lines.
The next thing to understand about the standard epidemiological account is that laboratory tests for Ebola disease only became available in West Africa on March 21st, 2014. Every step in the infection chain in figure 2 (except when C12 infected C14) relies on symptoms alone. Thus even the solid lines in figure 2 are highly provisional too.
The third important consideration is that there are two alternate versions of Baize et al. 2014 available online. The version initially published by the NEJM is represented by figure 2. The version available now (dated July 12, 2022) on the NEJM website states that the first (peer-reviewed) version of the article was “preliminary”. The major change in the final text was to state that a second epidemiological investigation concluded death dates for S1 (Emile), S2, S3 and S4 were later by up to 3-4 weeks (Baize et al., 2014). However, no information about this second investigation is provided. There is no explanation for why it was necessary and it contradicts figure 2 (which remains unchanged in the new version of the paper). It is also never stated which of the two conclusions, if any, the authors themselves favour.
(Potentially, this second investigation was the one conducted by the Leendertz team and noted in Saéz et al., 2014. These researchers claimed that the first Baize investigation was incorrect and that Emile (S1) died on Dec 28th (not December 6th) and his sister on January 5th (not December 29th) and his mother (S3) on January 11th (not December 13th). However, Saéz et al. do not mention revising the death date of the grandmother (S4) which Baize et al. state was revised in the second investigation from January 3rd to an unspecified date.)
This epidemiological uncertainty manifests even at later dates and among confirmed cases. For example, figure 2 does not include patient C3, who was the first confirmed Ebola death (on March 17th) at Guéckédou hospital. This is the nearest hospital to Meliandou, 12 km away. Nor was a chain established for one of the next two deaths (C4 on March 18th) at Guéckédou hospital (both are therefore only noted in Table 1 of Baize and not figure 2).
This insufficiency of concrete data surrounding the very early cases, plus the lengthy time interval separating Emile’s death in December from the first confirmed cases in mid-March and the delayed subsequent investigations cast considerable doubt on the validity of the whole chain.
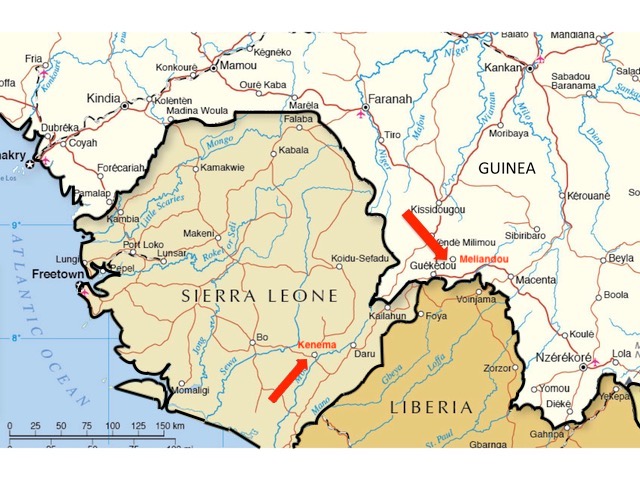
There are other problems too. As can be seen in figure 2, if we consider only the first cases confirmed by laboratory tests, the earliest deaths were not recorded at Guéckédou hospital at all. Despite being labeled with higher numbers (as if they were later in the outbreak), the four earliest deaths among confirmed cases were C12 (d. Feb 28th), C13 (d. March 12th), C8 (d. March 16th), and C14 (d. March 16th). These patients all presented at Macenta hospital which is over 100 km east of both Meliandou and Guéckédou (see figure 1, above).
This pattern among the earliest confirmed deaths does not obviously suggest an epicentre in Meliandou. So, although Baize et al. conducted epidemiological investigations, this rooting of the outbreak is, in reality, highly provisional.
Last, but perhaps not least, since the diagnoses are mostly unconfirmed, the credibility of the epidemiological chain in figure 2 rests heavily on the researchers’ claims to have interviewed eyewitnesses, yet Emile’s father contradicted them in 2015, telling Reuters:
“It wasn’t Emile that started it”.
Zaire Ebola in 2014: the prima facie case for a lab origin
After Ebola was first confirmed by laboratory tests in mid-March 2014, persistent rumours in the region linked the outbreak to a US-run research laboratory in Kenema, Sierra Leone (Wilkinson, 2017). This facility studies viral hemorrhagic diseases, of which Ebola is one.
Kenema is 140 km southwest of Guéckédou and the same distance from Macenta and a little farther from Nzérékoré (see figure 1, above). All these towns sit close to or fairly close to the border where Guinea, Sierra Leone and Liberia meet.
The Kenema laboratory stands in the grounds of the Kenema Government Hospital (KGH). The lab, but not the hospital, has been operated by the US-based Viral Hemorrhagic Fever Consortium (VHFC) since 2010. Though there is an interesting continuity–a viral hemorrhagic fever laboratory was run by the US CDC in Kenema between 1976 up to Sierra Leone’s civil war in the 1990s.
The president and founder of the VHFC is virologist Robert (Bob) Garry of Tulane University.
In response to lab origin speculation, Garry gave a curious denial of any connection to his lab in an interview with Politifact published on 15 October, 2014, titled “5 falsehoods about Ebola“. Garry told his interviewer, Aaron Sharockman:
“We were there working 10 years and then Ebola came here.”
The possibility of a lab origin never gained much attention outside Africa. But the 2014 Ebola outbreak was enigmatic on several key grounds.
One puzzle was noted by the Leendertz group:
“[The] current epidemic represents the first proven emergence of Zaire Ebola virus in West Africa.” (Saéz et al., 2014)
The Zaire species of Ebola is the most lethal (to humans) of all the members of the Ebola genus. This genus also includes other Ebola-like viruses: Bundibugyo virus, Tai Forest Virus, Reston Ebola, Bombali Ebola, and Sudan Ebola. As its name implies, all prior outbreaks of Zaire Ebola were in the central African equatorial zone (the Congo basin) (Feldmann and Geisbert, 2011). At its closest, this classical Zairean Ebola zone is 3,000 km from Guinea. Hence Zaire Ebola’s appearance in West Africa was a striking and very unexpected development.
How might it have reached West Africa? Ebola is not highly contagious. Transmission normally requires direct contact with the body fluids of an infected host. With such weak infectious properties and poor spreading potential, how could it move so far? Moreover, although often lethal and hence relatively easy to spot when it emerges, the virus caused no known human or animal outbreaks en route from its traditional Congo refuge.
A second major puzzle is that subsequent genome sequencing and phylogenetic analysis has shown unambiguously that the 2014 outbreak resulted from a single jump into humans (Gire et al., 2014; Dudas and Rambaut, 2014).
Zoonotic outbreaks, including most past Ebola outbreaks, typically feature multiple jumps to humans from an animal source (Feldmann and Geisbert, 2011). Single jumps, however, are consistent with lab origins and are often considered a red flag for that possibility (Nakajima et al., 1978). The reason is that researchers often work with a single isolate, perhaps one that they have found is particularly easy to replicate in the laboratory, whereas natural populations are typically diverse. This difference provides a genetic signal for distinguishing natural origins from laboratory ones.
Last, Zaire Ebola is the species favoured by civilian and military research labs for studying Ebola-type viruses. It is their focus because of its high mortality rate and thus biowarfare potential.
Journalist Chernoh Bah is now a graduate student at Northwestern University. Noting the gap between the weakness of the Leendertz account of the outbreak origin (and we would add that of Baize too) and the forcefulness with which the Emile narrative was asserted by western scientists and western media, he wrote:
“it is difficult not to interpret the ‘zoonotic origin of the West African Ebola epidemic’ narrative advanced by Fabian Leendertz and his team as part of a cover-up or obfuscation of the actual chain of events that laid the foundation for the West African Ebola outbreak.” (Chernoh Bah, The Ebola Outbreak in West Africa)
Indeed, the Kenema laboratory merits close inspection as a potential source of the Zaire Ebola strain that led to the 2014 outbreak.
Did the Viral Hemorrhagic Fever Consortium study Ebola at Kenema?
According to its website, the Viral Hemorrhagic Fever Consortium is a collaborative project for the study of hemorrhagic fevers and their treatments. Lassa virus, which is common locally, and the Ebola viruses are the most prominent viral hemorrhagic diseases. The consortium’s only permanent site is at Kenema.
Prominent institutional members of the VHFC include labs from Harvard University, the Broad Institute of MIT and Harvard, Scripps Research of San Diego, and Zalgen, a diagnostics company founded by Robert Garry. Though not currently listed as such, in 2014, Metabiota, the self-described “pandemic threat management” company was a VHFC partner as have been other companies.
Like Garry, the other VHFC leadership is US-based and includes Kristian Andersen of Scripps Research, Erica Ollmann Saphire, and Pardis Sabeti (Board Treasurer). Garry and Andersen are currently highly visible due to their numerous media appearances and scientific articles dismissing SARS-CoV-2 lab-origin hypotheses as baseless conspiracies (e.g. Andersen et al., 2020; Worobey et al., 2022).
In 2011, three years before the West African Ebola outbreak, Reuters profiled the research in Kenema at length. Readers were told that a “laboratory in southeastern Sierra Leone is an outpost of the U.S. government’s ‘war on terror,’ funded by a surge in bio-defense spending since the airplane and anthrax attacks on New York and Washington a decade ago. American research aims to limit the vulnerability of western interests to biological agents.”
As Reuters noted: “In 2001 — prior to the September 11 attacks — the U.S. National Institutes of Health budget for bioterrorism and related research was $53 million. But by the fiscal year 2007 the NIH was requesting more than $1.9 billion.” Reuters concluded that the Kenema labs’ share of that allocation was $40 million.
On August 25, 2013, just months before the Ebola outbreak, the VHFC posted on its website an article titled: “Researchers at the Scripps Research Institute make major advances in the fight against Ebola virus.” This article was later removed but its existence is verifiable using the WayBackMachine. Nevertheless, the title alone raises some key questions: Why did the VHFC post about Ebola if it wasn’t working on it at the time? In particular, what Ebola variant was being studied? What was the nature of the experiments? Why remove the post?
This was not the only time the VHFC appeared to be steering perception of its research towards Lassa fever and away from Ebola in the wake of the outbreak. On May 27th, 2014, a VHFC statement appeared that referred to the “current Ebola Epicenter of Guéckédou, Guinea” and the collaboration with Metabiota. The statement referenced their “Lassa laboratory at Kenema Government Hospital (KGH)”. This is the first reference we have been able to find of the VHFC calling its facility at Kenema the “Lassa Laboratory”. This may be an understandable rhetorical effort to distance themselves from Ebola research. But it raises the question: if the work at Kenema was only on Lassa, why was the generic term “hemorrhagic fever” used for the consortium (and previously the lab), if not to encompass research on related viruses? And were such large funding amounts simply for Lassa fever?
We do know that Ebola was important to the VHFC and its partners and a primary interest for at least some of its members.
Indeed, all the leading US-based researchers of the VHFC, Robert Garry, Kristian Andersen, Erica Ollmann Saphire and Pardis Sabeti have published multiple original research papers on Ebola virus (e.g. Lee et al., 2008; Koehler et al., 2013; Murin et al., 2014; Guha et al., 2018; Gunn et al., 2018; Barnes et al., 2020).
An Ebola focus also accords with US biosecurity research priorities under whose auspices the Kenema lab is largely funded. While Lassa fever is an endemic and sometimes debilitating illness, Ebola is a mysterious but highly lethal pathogen associated with the very highest level of biosecurity concerns.
Two exemplars are of this personal interest in Ebola are Erica Ollmann Saphire and Thomas Geisbert.
Erica Ollmann Saphire
Erica Ollmann Saphire is a VHFC board member from the La Jolla Institute for Immunology in San Diego. Saphire’s main research focus is Ebola. In January 2013, for instance, a lengthy and hyperbolic profile of her was published by Gary Robbins of the San Diego Union-Tribune: “The Virus Hunter: Erica Ollmann Saphire takes on deadly threats.”
The piece begins: “Deep in the thicket of west Africa, on a bamboo bridge strung over raging waters, Erica Ollmann Saphire groped through the dark toward a village where pestilence can snuff out life with ruthless efficiency. She was looking for rodents. The Scripps Research Institute biologist wanted to know how and where her enemy spreads viral hemorrhagic fever: things like Ebola and Lassa, diseases that can kill.”
The piece notes that Saphire was working on a “species of Ebola” which is “50-90 percent lethal.” This part is not hyperbole–since 2006 Saphire has co-authored over 30 publications on Zaire Ebola virus, testament to a longstanding research interest (e.g. Bornholdt et al., 2013).
Thomas Geisbert and the Failed Clinical Trial of TKM-Ebola
Another example is Thomas Geisbert. Geisbert is now at the University of Texas Medical Branch, but formerly he was at the United States Army Medical Research Institute of Infectious Disease (USAMRIID) in Maryland, also known as Fort Detrick. It is the premier US “biodefense” facility.
On Dec. 14, 2014, Andersen published a post on the VHFC website celebrating Time magazine’s choices of “VHFC researchers Drs. Pardis Sabeti and Thomas Geisbert” as “Ebola Fighters” and two of their people of the year.
Geisbert’s 2014 Time profile is informative. Citing Soviet defector Ken Alibek as his authority (even though Alibek was, by then, widely considered an unreliable source), Geisbert told Time readers that the Soviet Union had tried to weaponize Ebola. While at Fort Detrick, Geisbert and his colleagues had pushed for a US response to Alibek’s allegations:
[at that period there] “wasn’t money or interest or time to take those products across the finish line.” “But after 9/11, everything changed. There was increased funding. It was fortunate for me, because Ebola was my main area of interest. When all the money became available, we started looking at developing a vaccine. The idea came from Heinz Feldmann, then at the Public Health Agency of Canada (now at the National Institute of Allergy and Infectious Diseases), and the first big success we had was in 2002 or 2003. We did two back-to-back studies, and this was the first vaccine that completely protected monkeys from Ebola.”
Geisbert’s research was detailed further by Constantine Nana, who wrote a book about the 2014 outbreak: The Ebola Outbreak in West Africa. In the book Nana wrote:
“[Geisbert] has studied the Ebola virus for more than two decades and spent several years working with the United States Army Medical Research Institute of Infectious Disease at Fort Detrick. In March of 2014, he was awarded (together with Profectus Biosciences, Tekmira Pharmaceuticals, and Vanderbilt University Medical Center) $26 million (to be distributed over five years) by the NIH to ‘advance treatments of the highly lethal hemorrhagic fever viruses Ebola and Marburg.’
In his book, Nana speculated that such research might have led to a leak at Kenema.
In particular, Nana cites a product called TKM-Ebola, an RNA-based therapeutic that, according to research published in the Lancet, was a 100% effective treatment for primates infected with Ebola (Geisbert et al., 2010). The Lancet work was funded by the Defense Threat Reduction Agency (part of the Department of Defense), it involved the Zaire Ebola strain, and was a collaboration involving Geisbert and Tekmira Pharmaceuticals.
In his Time magazine profile, Geisbert heralded the success he and Tekmira were allegedly having:
“I’ve got shelves and shelves and shelves and shelves of stuff that slow or inhibit growth of Ebola in culture. Dozens of those protect mice or guinea pigs. Only two worked in nonhuman primates, out of all the studies done in the BSL4 lab — and those are ZMapp done by Gary Kobinger in Canada and TKM-Ebola, which I worked on with biotech company Tekmira. … When you do have success, like when we did the TKM-Ebola study, there is no greater feeling.”
Based on the Lancet result, Tekmira raised at least $140 million from military-related funders. This allowed them, around the time of the outbreak, to run a series of Phase I and Phase II clinical trials of different versions of TKM-Ebola.
For instance, on Dec. 13, 2013, Tekmira announced a study in humans and on Jan. 14, 2014, wrote a further press release, titled “Tekmira Doses First Subject in Human Clinical Trial of TKM-Ebola”. As reported by the New York Times in June 2015, at least one phase II trial was definitely conducted in Sierra Leone. Funded by the UK’s Wellcome Trust, this one was halted because, according to Tekmira, the drug was “not likely to demonstrate an overall therapeutic benefit”.
This trial was later published in a scientific journal and described as occurring at Port Loko, 190 km northwest of Kenema (Dunning et al., 2016).
From the limited descriptions available, one of these trials fits the timing required for it to have triggered the 2014 Ebola outbreak but none of them fits the location. However, the data is incomplete; for his book, Constantine Nana corresponded with the lead investigator in the Port Loko Phase II trial, Dr. Peter Horby of the University of Oxford.
Horby told Nana “he had no information as regards the results of the Phase I trial.” To lead a Phase II trial and know nothing about that product’s Phase I trial is indeed mysterious and rather strange.
Soon after, in July 2015, Tekmira changed its name and ended its quest for an Ebola treatment.
At a minimum, these activities demonstrate the very active nature of Geisbert’s interest in Ebola virus and cause one to wonder about the sudden ending of Tekmira’s interest in Ebola after such a promising beginning. And it further raises the possibility that experiments might have been conducted at Kenema that did lead to an outbreak. For instance, perhaps live Ebola virus from unpublished sources was cultured alongside other reagents for these trials?
Other VHFC members
Pardis Sabeti, one of Time magazine’s “Ebola fighters” is a prominent researcher at the Broad Institute and is VHFC’s treasurer. A press release from the Vilcek Foundation on Jan. 29, 2014 states that she:
“has studied several agents that cause infectious diseases, such as the malaria parasite, Lassa virus, and Ebola virus.”
In 2013 Robert Garry co-authored a paper on a novel treatment for Zaire Ebola (Koehler et al., 2013). All eleven other authors were from USAMRIID, aka Fort Detrick. This site is the largest “biodefense” facility in the world and Garry’s company, Zalgen, is located close-by.
Other commercial research at the Kenema laboratory
In 2010, Corgenix, another “partner” of the VHFC, published a news release: “Corgenix Awarded NIH Grant to Develop Next Generation Technology Detection Products for Ebola and Marburg Viruses.” The release stated: “Collaborating with Corgenix on the study will be Tulane University, The Scripps Research Institute and Autoimmune Technologies, LLC.” Autoimmune Technologies has also been a VHFC partner.
“We expect this study will result in specific, cost-effective and easy to use tests for Ebola and Marburg virus detection,” said Jon Geske, Ph.D., Corgenix Project Director and Principal Investigator of the program. “In addition, the resulting diagnostics will be critical for development of vaccines and other treatments for these currently incurable diseases.”
“Building on our very successful Lassa virus program, this will enable the development of state-of-the-art diagnostic tests for Ebola and Marburg viruses on multiple delivery platforms.” added Douglass Simpson, Corgenix President and CEO.
The 2010 Corgenix news release also quotes VHFC President Robert Garry:
“We have been very pleased with the results of our collaborative effort over the past five years. The diagnostic products for Lassa have shown to be remarkably effective in clinical settings in Africa and will have a meaningful impact on the healthcare in that part of the world, and will also fill a critical gap in bioterrorism defense. Now, under the new NIH grant, we will expand this program to address these additional infectious agents that have the potential to kill hundreds of thousands of people and are of concern to the public health and bioterrorism preparedness communities.”
This represents a further example of potentially highly relevant research (since to detect Ebola one usually needs Ebola) being conducted with members of the VHFC, quite possibly at Kenema.
Biosafety lapses at the Kenema lab
Studying viral hemorrhagic fevers is considered dangerous work. In the U.S., using live filoviruses requires biosafety level four (BSL-4) facilities, where researchers wear positive pressure ‘space suits’.
But in Kenema, also according to Reuters, biosafety “measures include goggles, gloves and masks.” The article quoted VHFC member Matt Boisen, a U.S. scientist from Tulane, now with Zalgen:
“Certainly we have less safety, less containment, but we do have the ability to do a lot more in the same amount of time.”
Apparently, lax biosafety protocols provided an incentive to work at Kenema.
In 2016 the Associated Press (AP) conducted a post-epidemic investigation into the Kenema lab and its role in the response. AP was told by an anonymous source that, at the Kenema lab, “used needles litter the place”.
AP also obtained internal WHO emails in which senior WHO official Pat Drury told WHO director Margaret Chan:
“Both labs [Tulane and Metabiota at Kenema] do not meet international standards for biosecurity.”
Others have corroborated this laxity. In the 2014 outbreak, the earliest emergency responder was the medical non-profit Doctors Without Borders (MSF) who were called in for their extensive Ebola experience.
MSF’s emergency response coordinator was Anja Wolz. She was highly critical of the biosafety measures used by Metabiota at Kenema. Having seen how they visited suspected Ebola cases, she told AP:
“I didn’t go inside the Metabiota lab…..I refused because I had already seen enough”
A CDC official, Austin Demby, later sent to investigate, reached similar conclusions. In an email about the Kenema lab he wrote:
“The cross contamination potential is huge and quite frankly unacceptable.”
Thus, there seems to have been a pattern at Kenema of lax biosafety procedures both before and during the outbreak.
MSF alleges a hidden outbreak in Sierra Leone
Responding to international emergency requests to attend an outbreak of then unknown cause, MSF arrived in Guéckédou, Guinea, on March 18th, 2014. It was their diagnostic efforts that first confirmed the presence of Ebola.
After the outbreak had waned, MSF wrote a report on the Ebola outbreak response that was highly critical of members of the VHFC.
The MSF report refers to their early suspicions that, despite the absence of positive test results from Sierra Leone, Ebola was nevertheless present:
“The detective work of the epidemiologists revealed some unconnected chains of transmission in different locations in the Guinée forestière region, many of whom had family in neighbouring Liberia and Sierra Leone.”
The report also cites Dr Armand Sprecher, MSF public health specialist:
“The problem initially was not so much the number of cases, but that the hot-spots were spread out in so many locations,”
In other words, the outbreak in Guinea did not resemble a recent outbreak with a simple epicentre, as proposed by the Leendertz and Baize papers. Instead, what MSF found when they arrived in Guinea were many cases, widely dispersed, at least some of which appeared to originate from neighbouring Sierra Leone.
This raised a further question:
“Meanwhile, there was concern all along about the puzzling absence of confirmed cases over the border in Sierra Leone.”
MSF had an explanation for this absence:
“From the onset of the epidemic, the U.S. biotechnology company Metabiota and Tulane University, partners of Sierra Leone’s Kenema hospital, had the lead in supporting Sierra Leone’s Ministry of Health in investigating suspected cases. Their investigations came back Ebola negative, while their ongoing surveillance activities seem to have missed the cases of Ebola that had emerged in the country.”
MSF’s suggestion that the Kenema lab was missing Ebola cases accords with AP’s investigation in Sierra Leone.
AP reporters obtained an email sent by WHO Ebola coordinator Philippe Barboza on 8th Aug, 2014. It stated:
[Metabiota staffers]“are systematically obstructing any attempt to improve the existing surveillance system and there are a lot of improvement(s) needed”
Another WHO official, outbreak specialist Eric Bertherat, had already reached similar conclusions. In an email dated July 17th he told colleagues there was “no tracking of the samples” and “absolutely no control on what is being done“. The result was “total confusion“. Later, a paper about the outbreak authored by Bertherat, Barboza, and others, referred to “considerable unmonitored transmission in the early months of the epidemic” and the withholding of data from the authors by Metabiota (Senga et al., 2017).
And Sylvia Blyden, a senior advisor to the Sierra Leone Government, told AP that Metabiota
“messed up the whole region.”
When MSF did at last get permission to work in Sierra Leone, they were taken by surprise:
“When we set up operations in Kailahun [a town equidistant between Kenema and Guéckédou], we realised we were already too late. There were cases everywhere, and we built the centre with 60 beds, rather than the 20 we started with in Guinea,” said MSF’s Anja Wolz.
But, just like the WHO, MSF got no cooperation from Kenema:
“The Ministry of Health and the partners of Kenema hospital refused to share data or lists of contacts with us, so we were working in the dark while cases just kept coming in.”
MSF agreed with Sylvia Blyden that failure at Kenema had major repercussions:
“After a short period of raised hopes in May as cases appeared to be declining in Guinea and Liberia, the hidden outbreak in Sierra Leone mushroomed and reignited the outbreak for its neighbours.”
This sentence is the second reference in MSF’s report to the Sierra Leone outbreak being “hidden”. Apparently, MSF thought that the failure to report cases from Sierra Leone was not a simple bungle.
These independent investigations, and the MSF report in particular, raise fundamental questions: Did the outbreak really begin in Guinea? Or did it in fact start in Sierra Leone? Did early testing and diagnostic failings in Sierra Leone bring about a narrative that placed the origin across the border in Guinea? Given the intentionality imputed by many of these witnesses to the failings in Sierra Leone, were they deliberate? If so, were they intended to divert attention away from the Kenema lab?
Phylogenetic analysis of the Ebola 2014 outbreak contradicts the standard narrative
To this day, the bat-tree-boy narrative still stands as the leading explanation for the West African Ebola outbreak of 2014 (Holmes et al., 2016). Even though no additional evidence in its favour has emerged since (such as the discovery of a natural source of Ebola in West Africa, despite intensive searching of wildlife), only very rarely do researchers even note that the story of Emile is effectively “anecdotal” (Spengler et al., 2016).
The 2014 Ebola outbreak (which is now referred to as the Makona strain of Ebola) marked the full debut of mass viral genome sequencing during an outbreak. In all, about 5 percent of confirmed Ebola cases in West Africa were sequenced, with each sequence having sampling time and location data attached to it. The total data set contains about 1,500 Ebola genomes, including from some of the earliest known cases.
Phylogenetic analysis is a set of methods to organise and visualise this vast quantity of genetic information. It can give detailed insights into the timing and connection between cases and so phylogenies have the potential to resolve where and when the virus emerged (see e.g. Holmes et al., 2016). For example, phylogenetic analysis has been used to show instances of the disease crossing and re-crossing the borders between Guinea, Sierra Leone and Liberia, and in which direction (Dudas et al., 2017).
This genetic information is now publicly available. It can be conveniently accessed and visualised via the open-source virology website Nextstrain.org, which is the source of the phylogenies in the figures below.
Since the on-the-ground epidemiology is clearly uncertain, the chief scientific reason for not taking the possibility of a lab origin seriously has been that a large number of phylogenetic analyses have been performed and these have consistently stated that the origin was unambiguously in Guinea (Dudas and Rambaut, 2014; Baize et al., 2014; Gire et al., 2014; Carroll et al., 2015; Hoenen et al., 2015; Kugelman et al., 2015; Ladner et al., 2015; Park et al., 2015; Simon-Loriére et al., 2015; Quick et al., 2016; Tong et al., 2016; Arias et al., 2016; Holmes et al., 2016; Dudas et al., 2017).
At first glance it seems clear why this should be. Figure 4 is a screenshot of the whole 2014 Ebola outbreak. It is taken from Nextstrain’s interactive Ebola phylogeny page. (Note: For this screenshot, and all the figures below it, specific Nextstrain settings were selected to highlight sampling dates and their country of origin.)
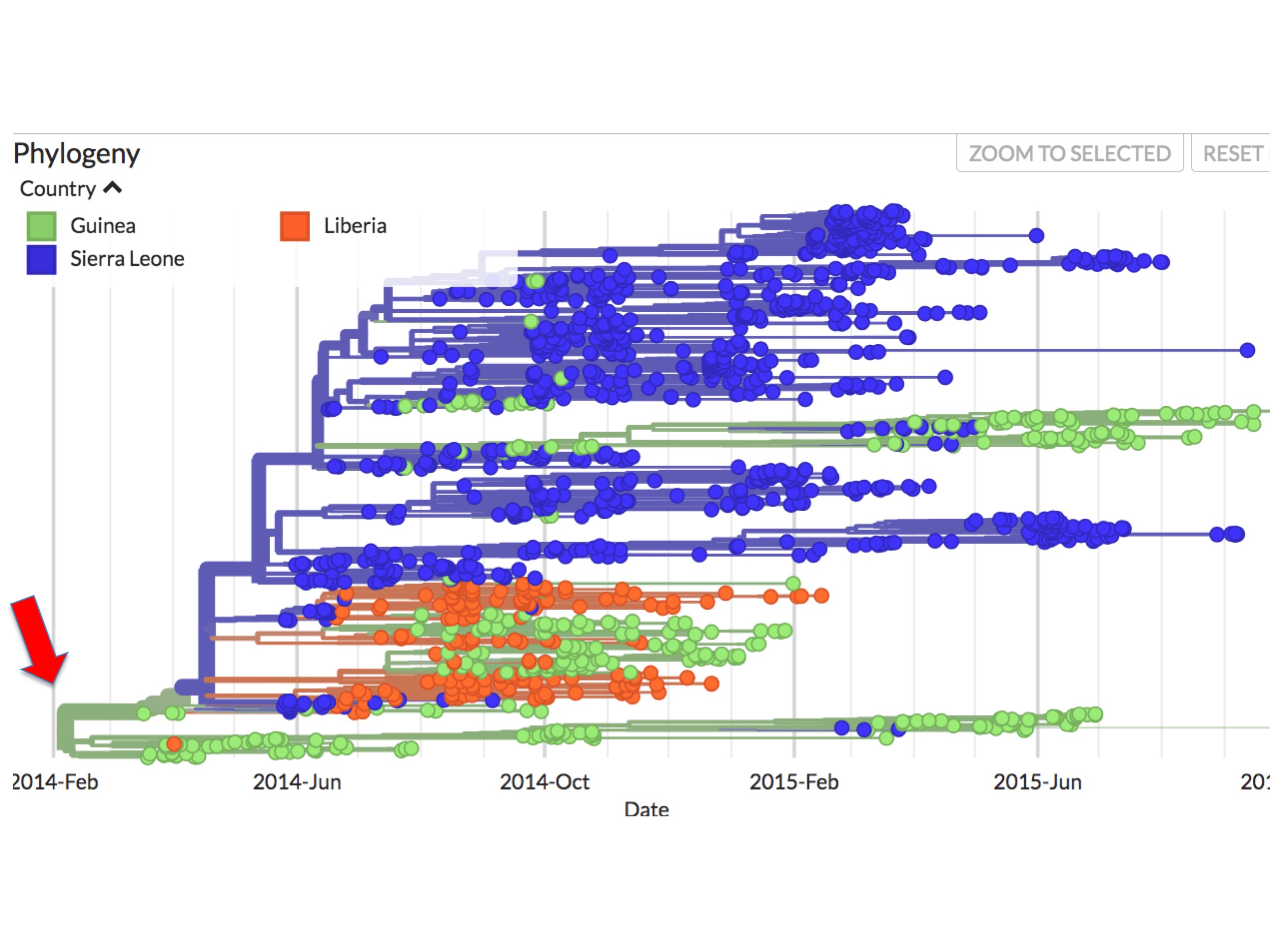
In Nextstrain (and thus figure 4 and succeeding figures), spots correspond to individual Ebola genomes sampled from single patients. Green spots are those diagnosed in Guinea; blue spots are patients diagnosed in Sierra Leone; and orange spots are cases from Liberia. The lines between them indicate inferred evolutionary connections between each sampled genome, resulting in a phylogenetic tree. The colours of these lines reflect an inference. They are Nextstrain’s calculation of the main country where the lineage is found.
In figure 4 it can be seen that all the early (prior to May 25th, 2014) sequences map to Guinea. Presenting the data this way implies that the root virus of the epidemic (red arrow at the left) jumped into humans in Guinea.
Though superficially simple, this interpretation gives rise to several anomalies, however.
Phylogenetic anomalies from assigning the root to Guinea
As figure 4 showed, beginning very early in the outbreak, the virus became separated into two major genetic lineages (i.e. branches).
This bifurcation can be better seen in figure 5. It is a screenshot of the same webpage but focusing only on the origin. It identifies two separate lineages called here SL-1 and GU-1.
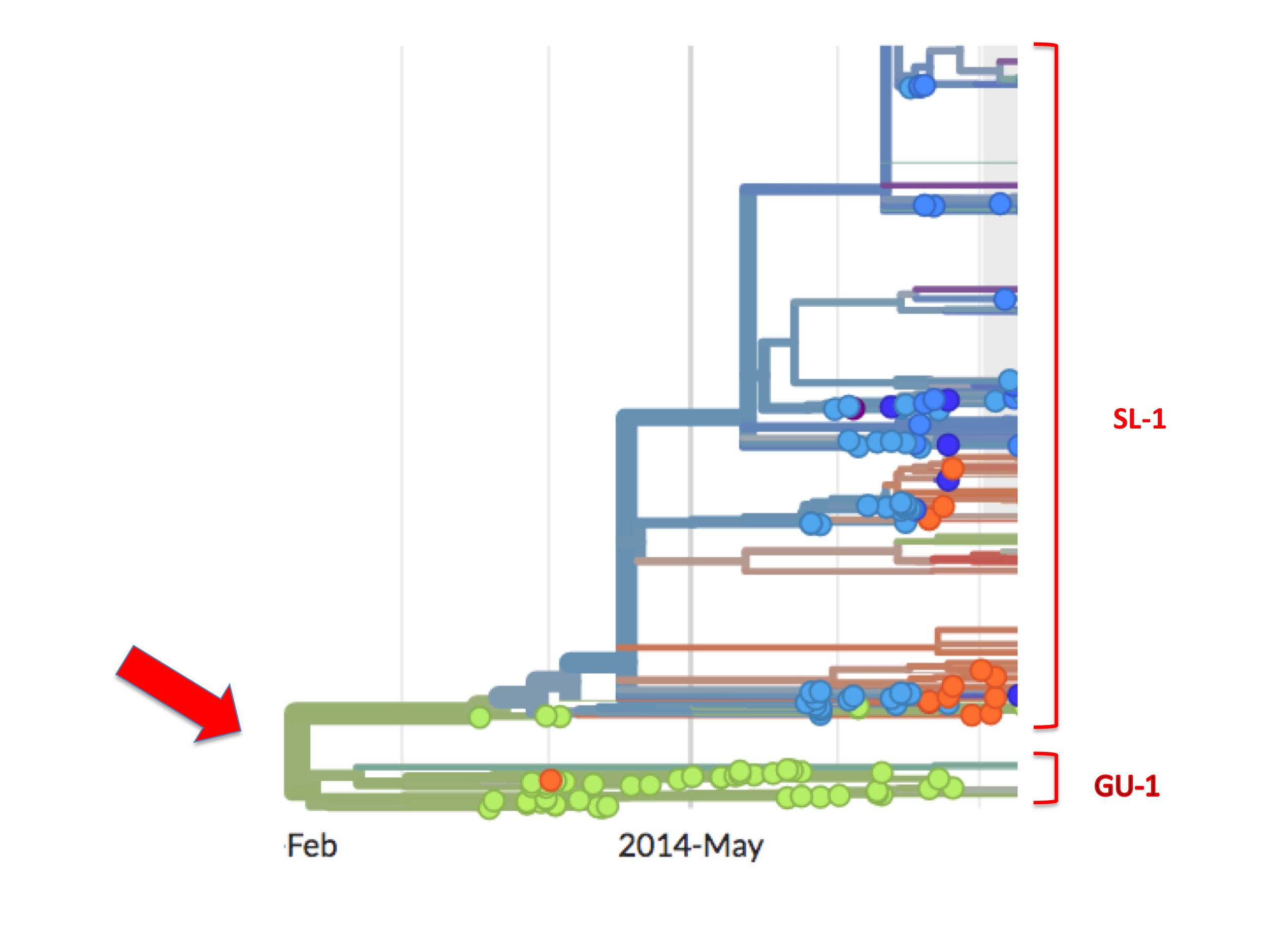
Of the two, the GU-1 lineage presents a straightforward case. This lineage arose in Guinea (the first examples are green spots) and most descendant viruses are also coloured green. That is to say, from its establishment to its extinction, the GU-1 lineage was almost entirely confined to Guinea. Of the few exceptions one is an orange (i.e. Liberian) spot in the GU-1 lineage. Presumably, one person infected with a GU-1 lineage virus travelled to Liberia where they were sampled and their genome was sequenced. There is also a small cluster of blue/purple spots dating from early 2015 (that are not visible in figure 5). A cluster implies, similarly, that one infected person took the GU-1 strain to Sierra Leone causing a small outbreak there and some of these descendants were diagnosed and sequenced.
A lineage beginning in Guinea and largely staying there is to be expected. Moreover, from Nextstrain one can see that the GU-1 lineage caused relatively few cases (compared to the SL-1 lineage). This concurs with the standard understanding that MSF brought the Guinea outbreak under control relatively quickly.
The upper lineage (SL-1) followed a very different pattern. First, it was not controlled and became responsible for the majority of cases in the entire West African outbreak. This lineage spread primarily in Sierra Leone but it also spread sporadically into Liberia (orange spots) and also into Guinea, as indicated by its green spots. SL-1 was thus responsible for most of the eventual 30,000 cases and most of the 11,000+ Ebola deaths.
However, the SL-1 lineage has a curious feature that is highlighted in figure 6.
SL-1 begins with three green (Guinean) spots (Kissidougou C-15, EM_079410 and EM_079442), but their viral descendants are all blue spots, meaning that these descendants were all Ebola cases diagnosed in Sierra Leone.
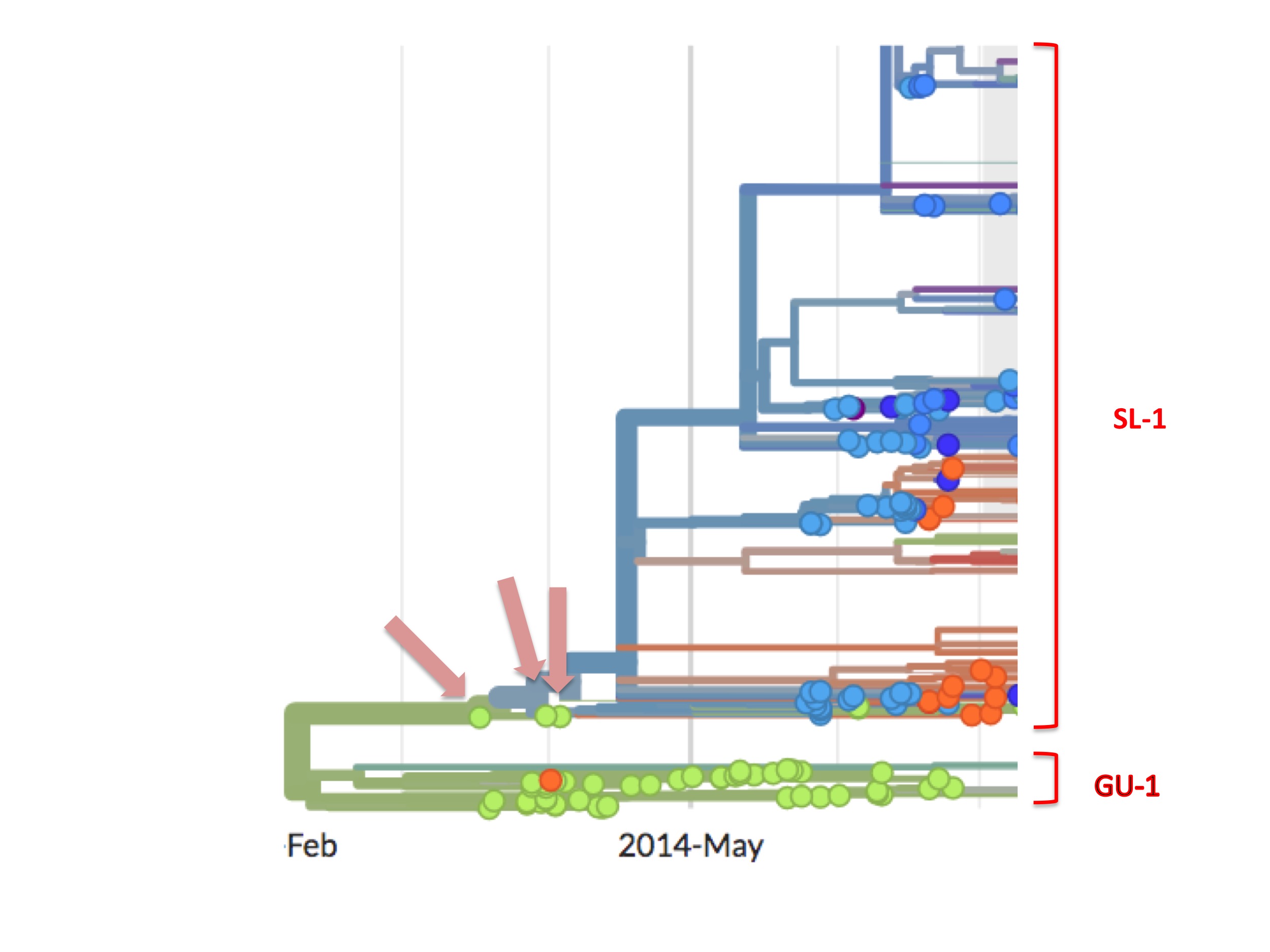
The orthodox interpretation of this has always been that the outbreak started in Guinea and spread into Sierra Leone.
However, looking carefully, this interpretation is difficult to sustain, for three reasons. First, from the data on the GU-1 lineage we know that a Guinean taking a GU-1 strain of the virus to another country was a rare event. It happened just twice in the 18 months of the outbreak there, once to Liberia and once to Sierra Leone. Dudas et al. asked this question in a different way and they concluded that, in the whole outbreak (which was doubtless much larger than the 30,000 recorded cases), only nine times did anyone with Ebola enter Sierra Leone from Guinea (Dudas et al. 2016). The orthodox interpretation nevertheless requires that, among the small handful of initial Guinean cases, one of them took the virus to Sierra Leone.
Second, the absence of green spots downstream of the first three indicates that these early Guinean cases failed to infect any further Guineans.
Thirdly, the disappearance of the SL-1 lineage in Guinea coincided in time with its appearance in Sierra Leone. Thus, the SL-1 lineage did not just spread into Sierra Leone, it also, simultaneously, vanished from Guinea
Each one of the three requirements of the orthodox view are, on their face, somewhat unlikely. Taken together, in science-speak, they represent a non-parsimonious explanation that suggests we might consider alternative ones.
(As an aside, close inspection of Nextstrain’s Ebola data shows there are occasional green (Guinean) spots in the SL-1 lineage and, later on, minor green outbreaks. One such virus is EM_79876, indicated by the black arrow in figure 7 below. In principle, any of these green spots in the SL-1 lineage might represent a continuation of a cryptic Guinean SL-1 outbreak. However, we can be fairly confident that EM_79876 [sampled June 5th, 2014], and others not shown, are instead independent later cross-border introductions from the outbreak in Sierra Leone. This confidence derives first from the long time intervals between identification of the first three green spots and the appearance of these latter samples [e.g. EM_79876 was sampled on June 5th, over two months after its last Guinean ancestor, which was sampled on April 3rd], and second because of the high genetic similarity of these downstream green samples to viruses identified in Sierra Leone. This interpretation is confirmed by Nextstrain. This link is to a screenshot showing in close-up how Nextstrain interprets the provenance of the earliest of these green spots in the SL-1 lineage [EM_79876 and EM_79880, both sampled June 5th 2014, and some others]. By showing these green spots as descending from one or more blue lines, Nextstrain’s algorithm is agreeing with our interpretation: that such green spots are all descendants of viruses found in Sierra Leone, thus they are introductions from there and not indicative of cryptic spread in Guinea).
Thus, to recap, the phylogenetic tree of the full epidemic indicates that the SL-1 lineage did not merely spread from Guinea into Sierra Leone but moved wholesale into it. What that means in practice is that all early patients in Guinea carrying viruses from the SL-1 lineage infected only individuals from Sierra Leone and no one from Guinea, their supposed home country.
Such anomalies should raise eyebrows of an even minimally curious researcher. Why should a root virus first identified in Guinea fail to initiate an outbreak in Guinea? Why should cases in Guinea start an epidemic in Sierra Leone? And why also should the disappearance of the SL-1 lineage from Guinea coincide in time with its appearance in Sierra Leone?
Resolving the rooting conundrum
There is a potential simple resolution to this conundrum. Let us hypothesise that the three green samples at the root of the SL1 outbreak were mislabelled or misallocated, and thereby wrongly assigned to Guinea instead of Sierra Leone.
If one re-labels each of them blue (i.e. Sierra Leonean) then the early stages of the pandemic phylogenetic tree resolve into two branches with no wholesale leap of one lineage into Sierra Leone. There was an origin, which, sometime before sampling started, split into two branches. One branch (GU-1) spread in Guinea and was largely contained; the other (SL-1) spread in Sierra Leone and was not controlled.
This hypothesis simplifies the scenario greatly, but is there a good reason for suggesting such misattributions? There is.
According to the contact tracing described in MSF’s report, some early cases in Guinea had no connections there but instead appeared to come from Sierra Leone (which is only 12 miles from Guéckédou). If true, though diagnosed in Guinea and therefore attributed to Guinea (and given green spots by Nextstrain), such patients are better understood as part of a Sierra Leone outbreak.
The hypothesis is thus that the three early green cases at the root of the SL-1 lineage are not properly from Guinea but instead represent spillovers from an undetected outbreak in Sierra Leone. These cases were thus labelled green only because they were picked up in Guinea, where sampling and contact tracing were effective.
Adjusting the tree accordingly provides a solution that accords with the evidence and is parsimonious since it resolves what is an otherwise highly perplexing state of affairs. However, there is one difficulty.
For any cases to have spilled from Sierra Leone in March, 2014, there must have been Ebola in Sierra Leone. Yet, officially, the first case was not diagnosed there until May 25th, 2014 (Goba et al., 2016). However, as the next section will show, there is strong phylogenetic evidence for such an outbreak.
Before discussing that evidence, it is imperative to note that assigning these three cases to Sierra Leone has important consequences for locating the origin of the outbreak. The sampling date of one of them (Kissidougou-C15) is March 17th. It is the earliest Ebola Makona genome ever sampled and the other two are also among the very earliest cases known. So, just as researchers have used these three samples to deduce that the origin was in Guinea, relabelling them shifts the presumed origin to Sierra Leone.
The MSF report provides additional clues as to which of the two countries was the true source of the epidemic. Drawing on distinct strands of evidence, MSF suggested that it was Sierra Leone since: (1) contact tracing showed that some very early cases came from Sierra Leone; (2) the outbreak in Guinea was mysteriously dispersed (and note also that Baize et al.’s early cases, though located in Guinea, were all at different points along the border with Sierra Leone); (3) when MSF set up in Kailahun (in Sierra Leone) the outbreak was already much worse in Kailahun than was the outbreak in Guinea; (4) the MSF report also suggested an outbreak in Sierra Leone was ‘hidden’ by poor or absent diagnostic procedures. The email from WHO’s Phillippe Barboza corroborates this by separately alleging that, in Kenema, Metabiota was “systematically obstructing” surveillance. A lack of surveillance and sequencing in Sierra Leone is important. It would suffice to explain why, if there was an outbreak in Sierra Leone that occasionally was spreading into Guinea, no Sierra Leone samples (blue dots) occur at the base of the SL-1 lineage.
The following section therefore addresses this key question: was there an early outbreak in Sierra Leone?
Further evidence of a hidden outbreak in Sierra Leone
Inspection of the genome sequences at the base of the SL-1 branch reveals another anomaly.
The first Ebola genome sequence officially from Sierra Leone is an accession called EM_095. It was obtained from a patient on May 25th, 2014. This date is over two months after the first confirmed Guinean cases were sampled. The next day (May 26th) two more patient samples were taken in Sierra Leone and later sequenced, these are Ebola accessions G_3677 and EM_096. The genome of G_3677 differs from EM_095 by four mutations, while EM_096 differs from EM_095 by seven mutations. Just one day later, on May 27th, Ebola accession G_3670 was obtained from another patient. It differs from EM0_95 by six mutations. The following day (May 28th), G_3679 was obtained. It also differs from EM_095 by six mutations. However, these six mutations are not the same six mutations as those by which G_3670 differs from EM_095. What this genome sequencing indicates is that substantial genetic diversity in the Ebola virus population already existed in Sierra Leone by the time the medics there first started testing.
This diversity has a simple meaning. It was not generated in four days; rather, it shows that mutations had been building up in an unsampled viral population. Thus a viral outbreak in Sierra Leone had existed for some considerable time before May 25th.
Subsequent sequencing efforts in Sierra Leone, which were much more comprehensive, uncovered yet more evidence of early diversity, thus confirming and extending this finding.
This high viral diversity existing prior to May 25th is apparent from the Nextstrain phylogenetic tree (see figure 8, below). It shows up as a set of bifurcating blue lines prior to May 25th. A bifurcating tree without spots (i.e. samples), as highlighted in figure 8 by the red oval, represents Nextstrain’s prediction of viral intermediates early in the growth of the SL-1 lineage, even though these were never sampled.
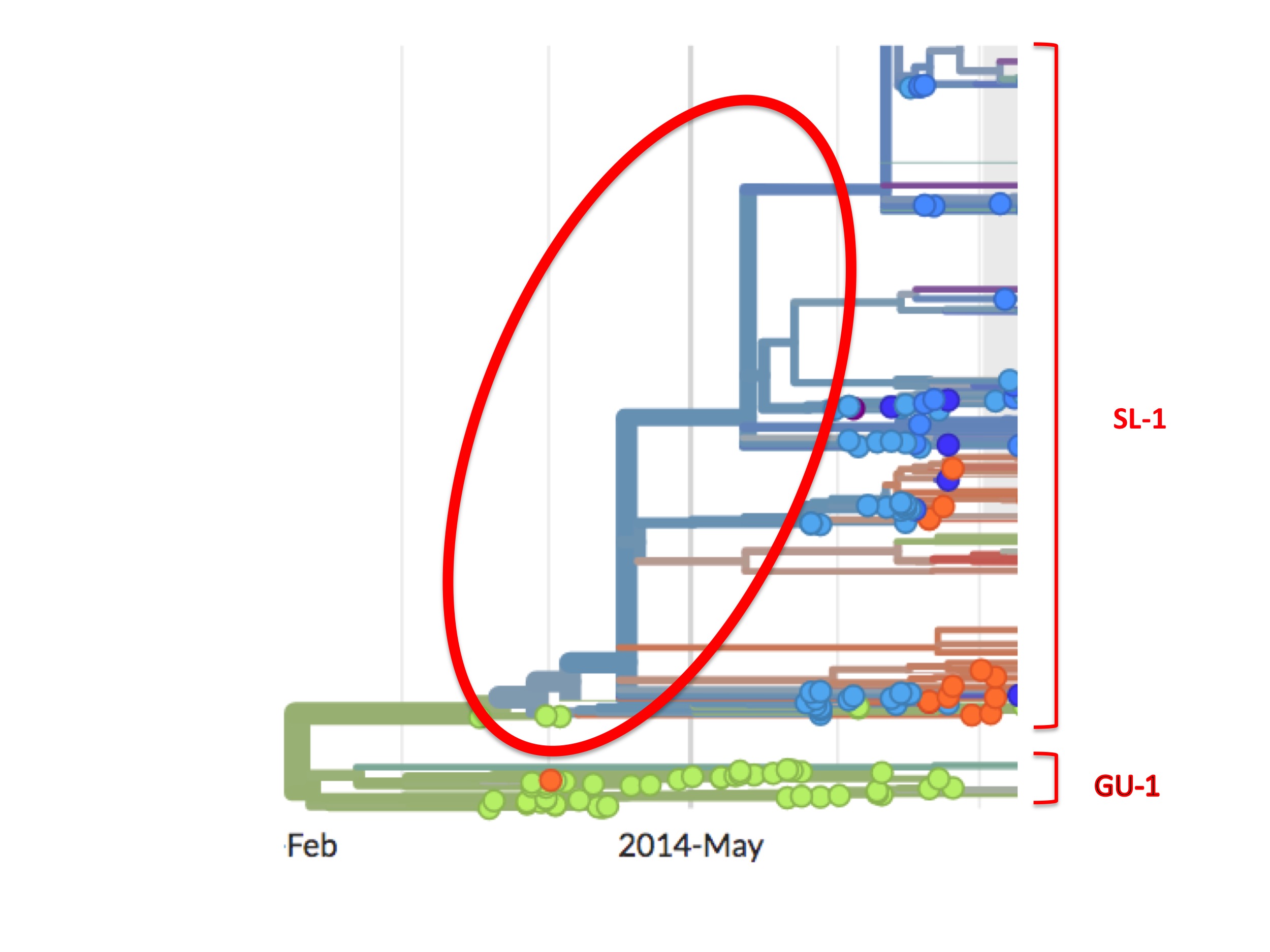
It is important to note that this diversity in the SL-1 lineage prior to late May was probably not from any outbreak in neighbouring Guinea at the time. This is because (as the figures also show) in Guinea there was abundant testing and sequencing (many green spots) prior to May 25th. If these or other virus strains had existed in Guinea they would likely have been picked up by the contact tracing and other efforts there, measures we know were comprehensive enough at that time to suppress the Guinea outbreak.
As noted above, the scientific literature contains dozens of papers detailing the events of the Ebola Makona outbreak. Of those published in high-profile journals, or in any of the fourteen papers presenting a phylogenetic analysis of the outbreak, there is no mention of these anomalies. However, three rarely-cited papers in low-profile journals (one is even in a “Supplement”) do acknowledge deficits in early sampling, contact tracing, and case ascertainment efforts in Sierra Leone, i.e. at the base of the SL-1 lineage (Wauquier et al., 2015; Goba et al., 2016; Senga et al., 2017).
Though in low-ranking journals, these last three papers are key data points since they represent researchers and epidemiologists in Sierra Leone corroborating MSF’s assessment of a missing or “hidden” outbreak in Sierra Leone.
The phylogeny: a summary
From the above it seems clear that the orthodox account of a Guinean origin for the Ebola 2014 epidemic is inconsistent on several counts with the accepted phylogenetic tree. One major anomaly is the unexplained early migration of the SL-1 lineage from Guinea into Sierra Leone. The second is the sudden and unexplained appearance in late May and early June 2014 of diverse Ebola strains in Sierra Leone that imply the existence of a substantial undetected outbreak there. However, these two anomalies are consistent with each other and with our proposed resolution, which is that the earliest confirmed case (Kissidougou C–15) and two others diagnosed in Guinea represent spillovers from Sierra Leone and not an origin in Guinea.
Furthermore, from MSF’s report and the WHO emails uncovered by AP, the epidemic is more likely to have begun in Sierra Leone and only later spread into Guinea.
This assemblage of evidence is interesting on several counts. Not only does it derive from diverse sources–phylogenetics, epidemiology, email correspondences–it also creates a coherent alternative account that fits all the data currently available. Taken together, the obvious inference is that the 2014 Ebola outbreak began in Sierra Leone and not Guinea.
A Sierra Leone origin is not per se a lab origin, however.
What is the origin of the Makona strain?
A major question nevertheless remains before considering the possibility of a lab origin–What is the source of the Makona strain?
The Makona strain of Ebola is not a standard or known strain, nor is it similar to any published strain. It is novel, having approximately 400 mutations that are not found in any previously known Ebola strain (Gire et al., 2014).
Hence, for the 2014 Ebola outbreak to have begun in a lab, the Makona strain must either represent the escape of an unpublished strain, perhaps one collected during fieldwork in central Africa. Alternatively, Makona could be a radically manipulated derivative of a known strain–either through genetic engineering or passaging. A combination of these two possibilities should also be considered.
Of these two alternatives, we know that Ebola and other viruses were being sought from wild animals in the Congo basin at the time as part of USAID’s PREDICT project. The chief actors in this were the Wildlife Conservation Society (WCS) and Metabiota, which, at the time, was at the time a partner of the VHFC.
Researchers from these two organisations searched the Congo basin and collected large numbers of blood samples, faecal samples and other genetic material from likely wild animal sources of Ebola: bats, animals captured for bushmeat, and apes (Olson et al., 2012; Reed et al., 2014; Seimon et al., 2015; Kumakamba et al., 2021). Other samples were obtained from human patients from the Congo (Grard et al., 2012). Some of this prospecting activity overlaps in time with the outbreak in West Africa.
Thus, one possibility is that Metabiota, or other collectors, used the VHFC lab at Kenema as part of a cold chain for the preservation of samples brought from the Congo basin. This might have been necessary since the Congo basin has historically been a politically unstable region and less friendly to US interests. The Kenema lab may also have been used for initial screening or testing of such samples. A third possibility is the formal or informal sharing of samples or strains with VHFC contacts or colleagues at Kenema, perhaps to help in the development of commercial treatments or diagnostic tools.
The kinds of samples that could have carried Ebola from the Congo basin include wildlife blood and tissue samples, samples from humans with suspected illnesses, and even whole animals or patients themselves. The possibility that the Kenema site was used for screening or testing of samples collected in other countries is especially intriguing in light of the statement that Matt Boisen made to Reuters: “we do have the ability to do a lot more in the same amount of time.”
Given these potentialities it is remarkable to discover that, in July 2014, during the epidemic, the VHFC wrote a brief report in which they accused Metabiota of an activity that would be riskier still. The VHFC accused Metabiota staff at Kenema of culturing cells from Ebola patients, which they insisted was dangerous and should “be stopped immediately.”
Metabiota issued a qualified denial, but the allegation is highly credible since the two organisations shared the same site; moreover its implications are very great. It suggests, first, that Metabiota had an interest in culturing novel strains of Ebola, second, that they had the technical capability and the personnel competent to do so at Kenema, and third, that they were willing to take exceptional risks. As the VHFC states, culturing Ebola in a small, uncertified, and unsecure lab would be very dangerous–culturing viruses is normally considered the most problematic biosafety step in virus research since it is an amplification process. The allegation therefore raises, in a very concrete way, the question of what Metabiota might have been doing in Kenema prior to the outbreak.
We know too, from the history of lab acquired infections of Ebola and Ebola-like viruses, that spillovers to humans do not require cultivation of the virus and can result from research accidents with live or deceased animals, with isolated tissues, or through the manipulation of patient samples (Luby et al., 1969; Emond et al., 1977; Formenty et al., 1999 ; Anonymous, 2004). Also important to note is that, in some of these cases, Ebola was not known to be present in the sample when the accident occurred.
Thus, given the research interests and the capacities of the VHFC lab in Kenema and its collaborators, it is a relatively simple matter to theorise how a novel strain of Ebola, like Makona, might have reached Kenema and then spilled over there during routine research activities.
Interesting too is the dual role of Metabiota. Besides collecting samples from the wild, Metabiota was also the company that, at least according to MSF and the WHO, obstructed or mishandled testing and diagnosis at Kenema and that Sylvia Blyden alleged “messed up the whole region.” If a research error on the part of Metabiota was the source of the strain (and Metabiota’s incompetence has been widely alleged), or even suspected to be, they would have had a strong incentive to also ‘bungle’ the identification of early cases and so obfuscate the origin.
Funding Cutoffs
It is also possible that the government of Sierra Leone suspected that Ebola came from the Kenema lab.
On July 23, 2014, in the midst of the Ebola outbreak, its Ministry of Health and Sanitation used its Facebook page (now deleted) to set out a series of orders. These were widely reported in the Sierra Leone media and were to “be effectual immediately”.
One injunction was for “Tulane University” (Robert Garry’s home institution) and the treatment center at Kenema to cease admitting new patients. “Tulane” was also instructed to leave the Kenema lab. Part four instructed the Kenema lab “to stop Ebola testing during the current Ebola outbreak.”
This last instruction is especially intriguing. Literally, it implies the lab was conducting research on the virus. However, the wording is ambiguous; quite possibly, given the circumstances, the lab was only being instructed to cease testing for Ebola. Either way, amidst a catastrophic epidemic it is a seemingly counterproductive but striking step to publicly close down a major international testing and treatment site.
Furthermore, just two weeks later, on Aug. 7, 2014, again in the midst of the outbreak, the U.S. government announced a similar decision. It cut funding to Tulane and the VHFC.
As related by Reuters: “The National Institutes of Health rejected a proposal from New Orleans-based Tulane University to renew the five-year contract which expires in November, according to a July 30 letter from NIH reviewed by Reuters. The expiring contract is worth $15 million.”
“NIH declined to comment on the decision,” citing “federal government procurement integrity rules.”
The Consortium was nevertheless able to secure other sources of funding. At the end of 2014, Corgenix received an infusion of over $800,000 from the Bill & Melinda Gates Foundation and the Paul G. Allen Family Foundation that it split with its partners.
On October 17, 2014, President Barack Obama named Ron Klain “Ebola czar”. Klain is now chief of staff in the Biden administration. The same day, the Obama White House instituted a “pause on funding for any new studies that include certain gain-of-function experiments involving influenza, SARS, and MERS viruses.”
The official statement connected the decision to recent biosafety incidents at Federal research facilities which had received a measure of media coverage. The New York Times reported on the move, noting that the pause: “made no mention of Ebola or any related filovirus.”
However, the timing of the announcement is suggestive that the research pause and the Ebola outbreak were connected.
Did researchers fix the phylogenetic analysis to cover up the origin of the 2014 Ebola outbreak?
Because so many Ebola genomes were sequenced during the 2014-2016 Ebola outbreak, a succession of major scientific papers appeared that analysed the epidemic in great detail from a phylogenetic perspective (Dudas and Rambaut, 2014; Baize et al., 2014; Gire et al., 2014; Carroll et al., 2015; Hoenen et al., 2015; Ladner et al., 2015; Park et al., 2015; Simon-Loriére et al., 2015; Quick et al., 2016; Tong et al., 2016; Arias et al., 2016; Holmes et al., 2016; Dudas et al., 2017). Many of these paid especial attention to the beginning of outbreak and to establishing the identity of the root virus and its geographic location.
All of them placed the outbreak origin unambiguously in Guinea. None of them seems to have considered how placing the root there generates the questions and anomalies discussed above.
A partial explanation for this conviction that the epidemic began in Guinea is that, in an outbreak situation, there are two standard methods to establish the identity of a root virus (Lyons-Weiler et al., 1998). Both methods begin by constructing an unrooted phylogenetic tree (which simply demonstrates the relatedness of all the viruses in the data set without assigning a root virus). To choose a root virus, one employs a genetic out-group in the form of one (or more than one) slightly more distantly related genome(s) on the theory that the root virus in an outbreak will be the one most closely related to the out-group(s). Most phylogenetic trees are generated with this method. The second method uses the timing of samples to infer the root virus. The reasoning is that the root virus will be (or be closely related to) the earliest viruses sampled (Drummond et al., 2006).
The latter method, since it relies on sample dating, has a major potential flaw: a susceptibility to sampling biases. Supposing the epidemic started in Sierra Leone but samples were taken there only long after sampling had begun in Guinea. In that case, a method reliant on sample timing would wrongly place the root virus in Guinea. This flaw is well known to phylogenetic experts (Liu et al., 2020; Kumar et al., 2021). As Kumar et al. stated it:
“Some methods also incorporate sampling times in phylogenetic inference, but they will automatically favor placing the earliest sampled genomes at or near the root of the tree. This fact introduces circularity in testing the hypothesis that the earliest sampled genomes were ancestral because sampling time is used in the inference procedure.”
To anyone knowing (or suspecting) that sampling preferentially began in one location only, in this case because MSF was initially invited only to Guinea or because Metabiota and Tulane bungled their response (as was widely alleged), then relying on sample dating to infer the root of the 2014 outbreak amounts to circular reasoning.
Every one of the 13 phylogeny papers listed above, however, used sample dating to locate the root virus. Only four of them also used an out-group method. Of these four, three did not explicitly test (or state) whether out-group rooting confirmed the dating result (Gire et al., 2014; Dudas and Rambaut, 2014; Holmes et al., 2016). For the fourth, this test was premature since only three genome sequences (all from Guinea) were available then (Baize et al., 2014).
In other words, even though many of these papers were published in the foremost scientific journals, like Science (Gire et al., 2014; Hoenen et al., 2015), Nature (Carroll et al., 2015; Simon-Loriére et al., 2015; Quick et al., 2016; Tong et al., 2016; Holmes et al., 2016; Dudas et al., 2017) and Cell (Park et al., 2015), their conclusions, that the epidemic began in Guinea are unsound due to this circularity and failure to make use of an out-group.
The circularity of relying on timing alone was surely known to the senior authors (and presumably to peer-reviewers also). Many of these authors must also have been aware of MSF’s report and the AP investigation. Why then was the obvious step of testing the Guinea root with an out-group skipped in all these papers? Was it because doing so would contradict a Guinean origin?
We are not in a position to remedy that omission. However, what we can say is that not corroborating the clearly flawed clock method with the obvious test is a very puzzling and troubling omission–all the more so since placing the origin in Guinea generates clear genetic and epidemiological anomalies.
Our conclusion, just from the phylogeny of the outbreak alone, is that, much like the epidemiological studies on Emile and Meliandou, the certainty with which researchers have placed the origin in Guinea is unwarranted. Much more likely, the true site of emergence was in Sierra Leone.
A close-knit band of conflicted researchers
The totality of the phylogenetic evidence, supporting Sierra Leone as the source, must be considered alongside all the other evidence relevant to the origin. As noted above, despite much searching, there is so far no evidence for an animal reservoir for Zaire Ebola in West Africa (Goldstein et al., 2018). The Makona strain’s sudden appearance in the region was thus unexpected and is still unexplained. Furthermore, the epidemiological investigations in Guinea and Sierra Leone were inconclusive and unconvincing. There was, however, a single spillover event, which is also consistent with a lab origin. And last, there was a research laboratory nearby that specialised in viral haemorrhagic fevers. The VHFC lab may or may not have housed Ebola viruses but it certainly had a dubious biosafety record.
All of the evidence, including the phylogeny, is therefore consistent with a lab origin. It is hence exceedingly difficult to understand why the 2014 West African Ebola outbreak has repeatedly been cited as a clear-cut example of a zoonotic outbreak.
Hence, like Chernoh Bah before us, the unacknowledged extreme contrast between the standard account and the evidence base compelled us to consider whether there was not, at some level, a concerted scientific effort to deflect attention from the VHFC and its lab in Kenema.
Some observations about the authors of these phylogenetic papers on the origin of the 2014 Ebola outbreak (and the epidemiological ones too) seem pertinent.
The first point is the pattern of substantial overlapping authorship amongst the papers that studied the epidemic and especially among the phylogenetic analyses that placed the origin in Guinea. Some of these overlaps are highlighted in Table 1.
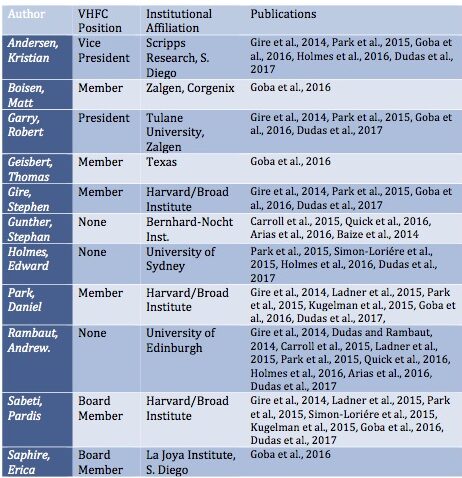
One or more of just six researchers are represented on all of them: Robert Garry, Andrew Rambaut, Stephan Gunther, Kristian Andersen, Pardis Sabeti, and Edward Holmes are lead authors on almost all of these publications. Of the two exceptions, one addresses only the Mali outbreak (Hoenen et al., 2015). The second is from China’s CDC (Tong et al., 2015). (However, a senior author of this paper is George Gao, China’s CDC recently departed head and a long-time associate of Rambaut and Holmes).
Second, many of the authors are now very prominent figures in the scientific disciplines of evolutionary virology and epidemiology (Andrew Rambaut, Robert Garry, George Gao, Edward Holmes, Gytis Dudas, Kristian Andersen, Wu-Chun Cao, Andreas Gnirke, Patrick Drury, Pierre Formenty, Trevor Bedford, Jonathan Towner, Gustavo Palacios, Stuart Nichol). One of them is celebrated geneticist Eric Lander, who, until he was forced to resign in February, 2022, was President Biden’s chief science advisor and is the founding director of the prestigious Broad Institute, whose Sabeti lab is a partner of the VHFC. (Gire et al., 2014).
Third, most of the senior authors of the phylogeny papers (notably, Robert Garry, Kristian Andersen, Pardis Sabeti, Erica Ollman Saphire, Daniel Park, and Stephen Gire) and plenty of less well-known authors, are directly connected to the VHFC and its Kenema lab. These authors in particular, have a career-sized conflict of interest, which they may also think is dwarfed by the possibility of being implicated in 11,000 deaths.
Lastly, in the face of widespread suspicions of a lab origin for SARS-CoV-2, many of these same authors (Robert Garry, Andrew Rambaut, Kristian Andersen, Edward Holmes, and Stuart Nichol) have become perhaps the most prominent and ardent defenders of a zoonotic origin for COVID-19.
How this overlap came about would seem a key question for both the Ebola and SARS-CoV-2 outbreaks.
As was learned from Freedom of Information Act requests, early in the COVID-19 pandemic, Anthony Fauci, head of the NIAID, secretly enlisted a small group of virologists to confer with (see p3134 of these emails) about whether SARS-CoV-2 originated from a lab. As evidence pointing to a lab origin for COVID-19 accumulated, this group evolved into what we have called Anthony Fauci’s COVID origin SWAT team. Not only were its members principals in arguing against lab origin theories, they even adopted many of the same scientific and phylogenetic misdirection strategies and tactics as those described above to suppress COVID-19 lab origin speculation.
Fauci’s initial discussion group included Drs Garry, Rambaut, Andersen, and Holmes, but what expertise did they bring to the table? Rambaut and Holmes had contributed to a handful of publications on the evolution of coronaviruses. On the other hand, Garry and Andersen, according to the standard database, Google Scholar, had never authored a single paper on coronaviruses before joining the group. Is it possible, therefore, that when COVID-19 broke out in Wuhan, the consideration uppermost in the mind of Anthony Fauci when he chose his secret circle was not scientific expertise but instead to find researchers familiar with the scientific and political challenges posed by a potential lab outbreak?
This link between Ebola 2014 and COVID-19 raises a closing question: to what extent is the COVID-19 outbreak, with its likely lab origin, a repeat event? Is COVID-19 the price to be paid for not conducting open, thorough, and forensic investigations of virus outbreaks, and instead leaving those tasks to the mercy of the researchers with the most vested interests?
Furthermore, it is difficult not to notice that the funding regime for pathogen research has spectacularly rewarded the researchers most involved in the Ebola 2014 outbreak. Though the VHFC and/or its then partner Metabiota are strong candidates to have initiated the whole event, and although they are also widely believed to have “messed up the whole region” by botching the initial response, the VHFC has profited greatly from the catastrophe. Not only did its personnel get to publish numerous papers in prestigious journals and so considerably enhance their careers, the VHFC now has a clinic three times the size of the old one. In 2016, the US Navy built a brand new clinic at Kenema with state of the art research, biosafety, water supply, and decontamination systems, including an entirely new building to serve as a ‘biorepository’ (Goba et al., 2016).
Such perverse rewards are forms of injustice. Like all injustice, they flourish in the dark. To serve justice, as well as to help prevent future outbreaks, the people of West Africa deserve to have the most intense light possible shed on the question of why Ebola came to them in 2014.
Sam Husseini is an independent journalist. Jonathan Latham, PhD, is a virologist.
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.
Arias, A., Watson, S. J., Asogun, D., Tobin, E. A., Lu, J., Phan, M. V., … & Cotten, M. (2016). Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Virus evolution, 2(1).
Baize, S., Pannetier, D., Oestereich, L., Rieger, T., Koivogui, L., Magassouba, N. F., … & Günther, S. (2014). Emergence of Zaire Ebola virus disease in Guinea. New England Journal of Medicine, 371(15), 1418-1425.
Barnes, K. G., Lachenauer, A. E., Nitido, A., Siddiqui, S., Gross, R., Beitzel, B., … & Sabeti, P. C. (2020). Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nature communications, 11(1), 1-10.
Bornholdt, Z. A., Noda, T., Abelson, D. M., Halfmann, P., Wood, M. R., Kawaoka, Y., & Saphire, E. O. (2013). Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell, 154(4), 763-774.
Carroll, M. W., Matthews, D. A., Hiscox, J. A., Elmore, M. J., Pollakis, G., Rambaut, A., … & Günther, S. (2015). Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature, 524(7563), 97-101.
Dudas, G., & Rambaut, A. (2014). Phylogenetic analysis of Guinea 2014 EBOV Ebolavirus outbreak. PLoS currents, 6.
Dudas, G., Carvalho, L. M., Bedford, T., Tatem, A. J., Baele, G., Faria, N. R., … & Rambaut, A. (2017). Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature, 544(7650), 309-315.
Drummond, A. J., Ho, S. Y. W., Phillips, M. J., & Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS biology, 4(5), e88.
Dunning, J., Sahr, F., Rojek, A., Gannon, F., Carson, G., Idriss, B., … & RAPIDE-TKM trial team. (2016). Experimental treatment of Ebola virus disease with TKM-130803: a single-arm phase 2 clinical trial. PLoS medicine, 13(4), e1001997.
Emond, R. T., Evans, B., Bowen, E. T., & Lloyd, G. (1977). A case of Ebola virus infection. Br Med J, 2(6086), 541-544.
Feldmann, H., & Geisbert, T. W. (2011). Ebola haemorrhagic fever. The Lancet, 377(9768), 849-862.
Forbes, K. M., Webala, P. W., Jääskeläinen, A. J., Abdurahman, S., Ogola, J., Masika, M. M., … & Sironen, T. (2019). Bombali virus in Mops condylurus bat, Kenya. Emerging Infectious Diseases, 25(5), 955.Gire, S. K., Stremlau, M., Andersen, K. G., Schaffner, S. F., Bjornson, Z., Rubins, K., … & Sabeti, P. C. (2012). Emerging disease or diagnosis?. science, 338(6108), 750-752.
Formenty, P., Hatz, C., Le Guenno, B., Stoll, A., Rogenmoser, P., & Widmer, A. (1999). Human infection due to Ebola virus, subtype Cote d’Ivoire: clinical and biologic presentation. The Journal of infectious diseases, 179(Supplement_1), S48-S53.
Geisbert, T. W., Lee, A. C., Robbins, M., Geisbert, J. B., Honko, A. N., Sood, V., … & MacLachlan, I. (2010). Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. The Lancet, 375(9729), 1896-1905.
Gire, S. K., Goba, A., Andersen, K. G., Sealfon, R. S., Park, D. J., Kanneh, L., … & Sabeti, P. C. (2014). Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. science, 345(6202), 1369-1372.
Goba, A. et al., (2016) An Outbreak of Ebola Virus Disease in the Lassa Fever Zone. The Journal of Infectious Diseases. Supplement 3 S110.
Goldstein, T., Anthony, S. J., Gbakima, A., Bird, B. H., Bangura, J., Tremeau-Bravard, A., … & Mazet, J. A. (2018). Discovery of a new ebolavirus (Bombali virus) in molossid bats in Sierra Leone. Nature microbiology, 3(10), 1084.
Grard, G., Fair, J. N., Lee, D., Slikas, E., Steffen, I., Muyembe, J. J., … & Leroy, E. M. (2012). A novel rhabdovirus associated with acute hemorrhagic fever in central Africa. PLos Pathogens e1002924.
Guha, S., Melnik, L., Garry, R. F., & Wimley, W. C. (2018). Ebola Virus Delta-Peptide Acts as an Enterotoxic Viroporin In Vivo. Biophysical Journal, 114(3), 265a.
Gunn, B. M., Yu, W. H., Karim, M. M., Brannan, J. M., Herbert, A. S., Wec, A. Z., … & Alter, G. (2018). A role for Fc function in therapeutic monoclonal antibody-mediated protection against Ebola virus. Cell host & microbe, 24(2), 221-233.
Hoenen, T., Safronetz, D., Groseth, A., Wollenberg, K. R., Koita, O. A., Diarra, B., … & Sow, S. (2015). Mutation rate and genotype variation of Ebola virus from Mali case sequences. Science, 348(6230), 117-119.
Holmes, E. C., Dudas, G., Rambaut, A., & Andersen, K. G. (2016). The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature, 538(7624), 193-200.
Koehler, J. W., Smith, J. M., Ripoll, D. R., Spik, K. W., Taylor, S. L., Badger, C. V., … & Schmaljohn, C. S. (2013). A fusion-inhibiting peptide against Rift Valley fever virus inhibits multiple, diverse viruses. PLoS neglected tropical diseases, 7(9), e2430.
Kugelman, J. R., Wiley, M. R., Mate, S., Ladner, J. T., Beitzel, B., Fakoli, L., … & National Institutes of Health. (2015). Monitoring of Ebola virus Makona evolution through establishment of advanced genomic capability in Liberia. Emerging infectious diseases, 21(7), 1135.
Kumakamba, C., Niama, F. R., Muyembe, F., Mombouli, J. V., Kingebeni, P. M., Nina, R. A., … & Lange, C. E. (2021). Coronavirus surveillance in wildlife from two Congo basin countries detects RNA of multiple species circulating in bats and rodents. PloS one, 16(6), e0236971.
Kumar, S., Tao, Q., Weaver, S., Sanderford, M., Caraballo-Ortiz, M. A., Sharma, S., … & Miura, S. (2021). An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic. Molecular Biology and Evolution, 38(8), 3046-3059.
Ladner, J. T., Wiley, M. R., Mate, S., Dudas, G., Prieto, K., Lovett, S., … & Palacios, G. (2015). Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell host & microbe, 18(6), 659-669.
Lee, J. E., Fusco, M. L., Hessell, A. J., Oswald, W. B., Burton, D. R., & Saphire, E. O. (2008). Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature, 454(7201), 177-182.
Leendertz, S. A. J., Gogarten, J. F., Düx, A., Calvignac-Spencer, S., & Leendertz, F. H. (2016). Assessing the evidence supporting fruit bats as the primary reservoirs for Ebola viruses. EcoHealth, 13(1), 18-25.
Leroy, E. M., Rouquet, P., Formenty, P., Souquiere, S., Kilbourne, A., Froment, J. M., … & Rollin, P. E. (2004). Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science, 303(5656), 387-390.
Leroy, E. M., Kumulungui, B., Pourrut, X., Rouquet, P., Hassanin, A., Yaba, P., … & Swanepoel, R. (2005). Fruit bats as reservoirs of Ebola virus. Nature, 438(7068), 575-576.
Leroy, E. M., Epelboin, A., Mondonge, V., Pourrut, X., Gonzalez, J. P., Muyembe-Tamfum, J. J., & Formenty, P. (2009). Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector-borne and zoonotic diseases, 9(6), 723-728.
Luby, J. P., and Sanders, C. V. (1969). Green monkey disease (” Marburg virus” disease): a new zoonosis. Annals of internal medicine, 71(3), 657-660.
Luebo, Democratic Republic of Congo, 2007. Vector-borne and zoonotic diseases, 9(6), 723-728.
Liu, Q., Zhao, S., Shi, C. M., Song, S., Zhu, S., Su, Y., … & Chen, H. (2020). Population genetics of SARS-CoV-2: disentangling effects of sampling bias and infection clusters. Genomics, proteomics & bioinformatics, 18(6), 640-647.
Lyons-Weiler, J., Hoelzer, G. A., & Tausch, R. J. (1998). Optimal outgroup analysis. Biological Journal of the Linnean Society, 64(4), 493-511.Murin, C. D., Fusco, M. L., Bornholdt, Z. A., Qiu, X., Olinger, G. G., Zeitlin, L., … & Saphire, E. O. (2014). Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proceedings of the National Academy of Sciences, 111(48), 17182-17187.
Nakajima, K., Desselberger, U., & Palese, P. (1978). Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature, 274(5669), 334-339.
Olson, S. H., Cameron, K., Reed, P., Ondzie, A., & Joly, D. (2012). Maximizing nonhuman primate fecal sampling in the Republic of Congo. Journal of Wildlife Diseases, 48(4), 888-898.
Park, D. J., Dudas, G., Wohl, S., Goba, A., Whitmer, S. L., Andersen, K. G., … & Sabeti, P. C. (2015). Ebola virus epidemiology, transmission, and evolution during seven months in Sierra Leone. Cell, 161(7), 1516-1526.
Pigott, D. M., Golding, N., Mylne, A., Huang, Z., Henry, A. J., Weiss, D. J., … & Hay, S. I. (2014). Mapping the zoonotic niche of Ebola virus disease in Africa. Elife, 3, e04395.
Quick, J., Loman, N. J., Duraffour, S., Simpson, J. T., Severi, E., Cowley, L., … & Carroll, M. W. (2016). Real-time, portable genome sequencing for Ebola surveillance. Nature, 530(7589), 228-232.
Reed, P. E., Mulangu, S., Cameron, K. N., Ondzie, A. U., Joly, D., Bermejo, M., … & Sullivan, N. J. (2014). A new approach for monitoring ebolavirus in wild great apes. PLoS Neglected Tropical Diseases, 8(9), e3143.
Saéz, A., Weiss, S., Nowak, K., Lapeyre, V., Zimmermann, F., Düx, A., … & Leendertz, F. H. (2015). Investigating the zoonotic origin of the West African Ebola epidemic. EMBO molecular medicine, 7(1), 17-23.
Seimon, T. A., Olson, S. H., Lee, K. J., Rosen, G., Ondzie, A., Cameron, K., … & Lipkin, W. I. (2015). Adenovirus and herpesvirus diversity in free-ranging great apes in the Sangha region of the Republic of Congo. PLoS One, 10(3), e0118543.
Senga, M., Koi, A., Moses, L., Wauquier, N., Barboza, P., Fernandez-Garcia, M. D., … & Lane, C. (2017). Contact tracing performance during the Ebola virus disease outbreak in Kenema district, Sierra Leone. Philosophical Transactions of the Royal Society B: Biological Sciences, 372(1721), 20160300.
Simon-Loriere, E., Faye, O., Faye, O., Koivogui, L., Magassouba, N., Keita, S., … & Sall, A. A. (2015). Distinct lineages of Ebola virus in Guinea during the 2014 West African epidemic. Nature, 524(7563), 102-104.
Spengler, J. R., Ervin, E. D., Towner, J. S., Rollin, P. E., & Nichol, S. T. (2016). Perspectives on West Africa Ebola virus disease outbreak, 2013–2016. Emerging infectious diseases, 22(6), 956.
Tong, Y. G., Shi, W. F., Liu, D., Qian, J., Liang, L., Bo, X. C., … & Cao, W. C. (2015). Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature, 524(7563), 93-96.
Walsh, P. D., Abernethy, K. A., Bermejo, M., Beyers, R., De Wachter, P., Akou, M. E., … & Wilkie, D. S. (2003). Catastrophic ape decline in western equatorial Africa. Nature, 422(6932), 611-614.
Wauquier, N., Bangura, J., Moses, L., Khan, S. H., Coomber, M., Lungay, V., … & Gonzalez, J. P. (2015). Understanding the emergence of Ebola virus disease in Sierra Leone: stalking the virus in the threatening wake of emergence. PLoS Currents, 7.
Wilkinson, A. (2017). Emerging disease or emerging diagnosis? Lassa Fever and Ebola in Sierra Leone. Anthropological Quarterly, 369-397.
Worobey, M., Levy, J. I., Serrano, L. M. M., Crits-Christoph, A., Pekar, J. E., Goldstein, S. A., … & Andersen, K. G. (2022). The Huanan market was the epicenter of SARS-CoV-2 emergence.




































































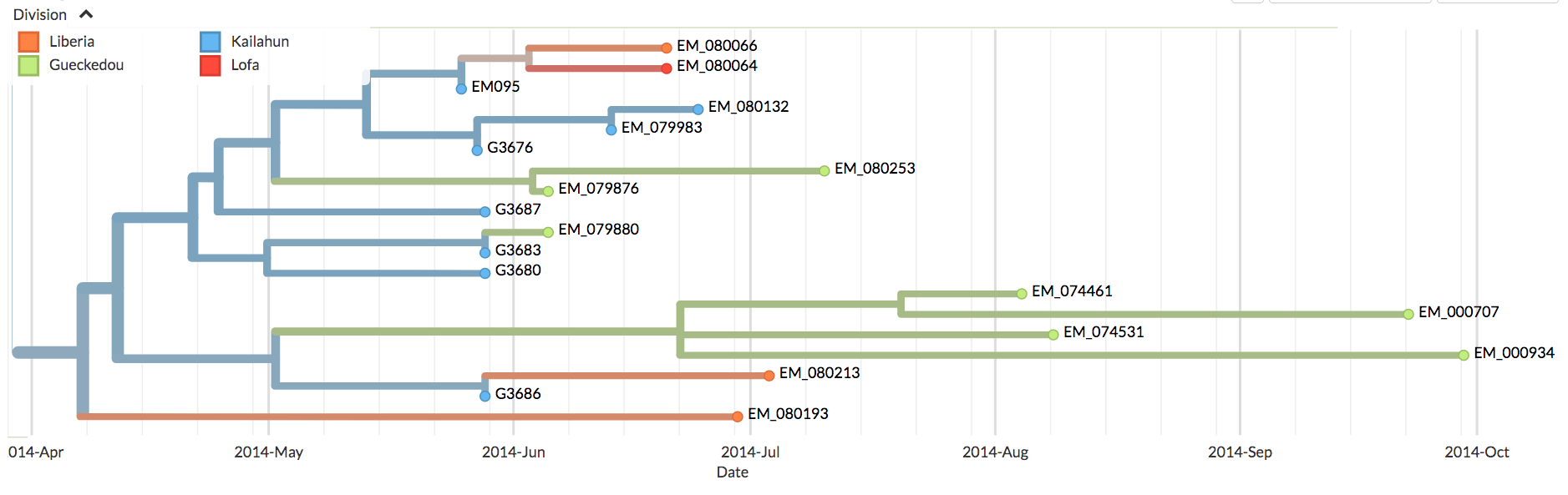{kind=link}
{kind=link}
{kind=link}
{kind=link}