Quote Dr. David Martin said Peter Daszak wrote in 2015 ‘they needed the media to hype up the need for coronavirus vaccines to sell them’. The very next year Dr Ralf Baric wrote in 2016: ‘..’the (WIV) virus was human ready’ a clear indication a weapon was being readied for human transmission. Seems to me J. Jyrkkanen ‘Collusion and Conspiracy to commit a crime for profit’.
Forwarded this email? Subscribe here for moreVSRF LIVE – Vax Mass Homicide With Special Guest, Denis Rancourt, PhDThursday, February 1: 7pm Eastern | 4pm Pacific Steve KirschFeb 1READ IN APPYou will NOT want to miss this! Tonight on VSRF LIVE I’ll be sitting down with Dr. Denis G. Rancourt, PhD. to discuss the COVID-19 vaccines’ potential role in wide-scale genocide occurring over the last 4 years.Dr. Rancourt is a former professor of over 20 years at the University of Ottawa, achieving the highest position of Full Professor until his departure in 2009. He has authored more than 100 articles published in leading peer-reviewed scientific journals in the fields of physics and environmental science. He is currently an independent social theorist and science critic covering the topics of medicine, COVID-19, individual health, climate change, geopolitics, civil rights, political theory, and sociology. Dr. Rancourt is an active volunteer with the Ontario Civil Liberties Association and also now collaborates closely with the registered non-profit “CORRELATION Research in the Public Interest” where he serves as Chair of the Board and an Associate Researcher.In 2020 Rancourt self-published the highly controversial, “All-cause mortality during COVID-19: No plague and a likely signature of mass homicide by government response” which postulated that COVID-19 vaccines and botched treatment protocols in public health settings were responsible for killing up to 17 million people worldwide. This research has been well received by many esteemed scientists and doctors worldwide and has truly blown open the COVID-19 vaccine and treatment debate on a global scale. We will be discussing this in detail.
Did we really need a vaccine or was there a drug out there that worked?
The above suggest the immune system was compromised but how?
A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence and study indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Repost w emphasis on spike used by Baric and Daszak.
Associated Revelation. Wuhan Institute of Virology Shao Cao admits they Tested Coronavirus Variants for the Best Adeherent to ACE2 Receptor
Abstract
The emergence of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome (MERS)-CoV underscores the threat of cross-species transmission events leading to outbreaks in humans. Here we examine the disease potential of a SARS-like virus, SHC014-CoV, which is currently circulating in Chinese horseshoe bat populations1. Using the SARS-CoV reverse genetics system2, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone. The results indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Additionally, in vivo experiments demonstrate replication of the chimeric virus in mouse lung with notable pathogenesis. Evaluation of available SARS-based immune-therapeutic and prophylactic modalities revealed poor efficacy; both monoclonal antibody and vaccine approaches failed to neutralize and protect from infection with CoVs using the novel spike protein. On the basis of these findings, we synthetically re-derived an infectious full-length SHC014 recombinant virus and demonstrate robust viral replication both in vitro and in vivo. Our work suggests a potential risk of SARS-CoV re-emergence from viruses currently circulating in bat populations.
Supplementary information
The online version of this article (doi:10.1038/nm.3985) contains supplementary material, which is available to authorized users.
Subject terms: Policy and public health in microbiology, Viral infection, SARS virus, Translational research
SPIKE PROTEIN CLOTTING PROBLEM
Dr. McCullough: The COVID Vaccines Are Causing the ‘Largest Blood Clots We’ve Ever Seen’
“My experience with these blood clots are they’re enormously resistant to blood thinners,” disclosed Dr. @P_McCulloughMD. “Wuhan spike protein is inside the blood clots, and it’s folding,… pic.twitter.com/UIawzXBToH
The emergence of SARS-CoV heralded a new era in the cross-species transmission of severe respiratory illness with globalization leading to rapid spread around the world and massive economic impact3,4. Since then, several strains—including influenza A strains H5N1, H1N1 and H7N9 and MERS-CoV—have emerged from animal populations, causing considerable disease, mortality and economic hardship for the afflicted regions5. Although public health measures were able to stop the SARS-CoV outbreak4, recent metagenomics studies have identified sequences of closely related SARS-like viruses circulating in Chinese bat populations that may pose a future threat1,6. However, sequence data alone provides minimal insights to identify and prepare for future prepandemic viruses. Therefore, to examine the emergence potential (that is, the potential to infect humans) of circulating bat CoVs, we built a chimeric virus encoding a novel, zoonotic CoV spike protein—from the RsSHC014-CoV sequence that was isolated from Chinese horseshoe bats1—in the context of the SARS-CoV mouse-adapted backbone. The hybrid virus allowed us to evaluate the ability of the novel spike protein to cause disease independently of other necessary adaptive mutations in its natural backbone. Using this approach, we characterized CoV infection mediated by the SHC014 spike protein in primary human airway cells and in vivo, and tested the efficacy of available immune therapeutics against SHC014-CoV. Together, the strategy translates metagenomics data to help predict and prepare for future emergent viruses.
The sequences of SHC014 and the related RsWIV1-CoV show that these CoVs are the closest relatives to the epidemic SARS-CoV strains (Fig. 1a,b); however, there are important differences in the 14 residues that bind human ACE2, the receptor for SARS-CoV, including the five that are critical for host range: Y442, L472, N479, T487 and Y491 (ref. 7). In WIV1, three of these residues vary from the epidemic SARS-CoV Urbani strain, but they were not expected to alter binding to ACE2 (Supplementary Fig. 1a,b and Supplementary Table 1). This fact is confirmed by both pseudotyping experiments that measured the ability of lentiviruses encoding WIV1 spike proteins to enter cells expressing human ACE2 (Supplementary Fig. 1) and by in vitro replication assays of WIV1-CoV (ref. 1). In contrast, 7 of 14 ACE2-interaction residues in SHC014 are different from those in SARS-CoV, including all five residues critical for host range (Supplementary Fig. 1c and Supplementary Table 1). These changes, coupled with the failure of pseudotyped lentiviruses expressing the SHC014 spike to enter cells (Supplementary Fig. 1d), suggested that the SHC014 spike is unable to bind human ACE2. However, similar changes in related SARS-CoV strains had been reported to allow ACE2 binding7,8, suggesting that additional functional testing was required for verification. Therefore, we synthesized the SHC014 spike in the context of the replication-competent, mouse-adapted SARS-CoV backbone (we hereafter refer to the chimeric CoV as SHC014-MA15) to maximize the opportunity for pathogenesis and vaccine studies in mice (Supplementary Fig. 2a). Despite predictions from both structure-based modeling and pseudotyping experiments, SHC014-MA15 was viable and replicated to high titers in Vero cells (Supplementary Fig. 2b). Similarly to SARS, SHC014-MA15 also required a functional ACE2 molecule for entry and could use human, civet and bat ACE2 orthologs (Supplementary Fig. 2c,d). To test the ability of the SHC014 spike to mediate infection of the human airway, we examined the sensitivity of the human epithelial airway cell line Calu-3 2B4 (ref. 9) to infection and found robust SHC014-MA15 replication, comparable to that of SARS-CoV Urbani (Fig. 1c). To extend these findings, primary human airway epithelial (HAE) cultures were infected and showed robust replication of both viruses (Fig. 1d). Together, the data confirm the ability of viruses with the SHC014 spike to infect human airway cells and underscore the potential threat of cross-species transmission of SHC014-CoV.
SARS-like viruses replicate in human airway cells and produce in vivo pathogenesis.
(a) The full-length genome sequences of representative CoVs were aligned and phylogenetically mapped as described in the Online Methods. The scale bar represents nucleotide substitutions, with only bootstrap support above 70% being labeled. The tree shows CoVs divided into three distinct phylogenetic groups, defined as α-CoVs, β-CoVs and γ-CoVs. Classical subgroup clusters are marked as 2a, 2b, 2c and 2d for the β-CoVs and as 1a and 1b for the α-CoVs. (b) Amino acid sequences of the S1 domains of the spikes of representative β-CoVs of the 2b group, including SARS-CoV, were aligned and phylogenetically mapped. The scale bar represents amino acid substitutions. (c,d) Viral replication of SARS-CoV Urbani (black) and SHC014-MA15 (green) after infection of Calu-3 2B4 cells (c) or well-differentiated, primary air-liquid interface HAE cell cultures (d) at a multiplicity of infection (MOI) of 0.01 for both cell types. Samples were collected at individual time points with biological replicates (n = 3) for both Calu-3 and HAE experiments. (e,f) Weight loss (n = 9 for SARS-CoV MA15; n = 16 for SHC014-MA15) (e) and viral replication in the lungs (n = 3 for SARS-CoV MA15; n = 4 for SHC014-MA15) (f) of 10-week-old BALB/c mice infected with 1 × 104 p.f.u. of mouse-adapted SARS-CoV MA15 (black) or SHC014-MA15 (green) via the intranasal (i.n.) route. (g,h) Representative images of lung sections stained for SARS-CoV N antigen from mice infected with SARS-CoV MA15 (n = 3 mice) (g) or SHC014-MA15 (n = 4 mice) (h) are shown. For each graph, the center value represents the group mean, and the error bars define the s.e.m. Scale bars, 1 mm.
To evaluate the role of the SHC014 spike in mediating infection in vivo, we infected 10-week-old BALB/c mice with 104 plaque-forming units (p.f.u.) of either SARS-MA15 or SHC014-MA15 (Fig. 1e–h). Animals infected with SARS-MA15 experienced rapid weight loss and lethality by 4 d post infection (d.p.i.); in contrast, SHC014-MA15 infection produced substantial weight loss (10%) but no lethality in mice (Fig. 1e). Examination of viral replication revealed nearly equivalent viral titers from the lungs of mice infected with SARS-MA15 or SHC014-MA15 (Fig. 1f). Whereas lungs from the SARS-MA15–infected mice showed robust staining in both the terminal bronchioles and the lung parenchyma 2 d.p.i. (Fig. 1g), those of SHC014-MA15–infected mice showed reduced airway antigen staining (Fig. 1h); in contrast, no deficit in antigen staining was observed in the parenchyma or in the overall histology scoring, suggesting differential infection of lung tissue for SHC014-MA15 (Supplementary Table 2). We next analyzed infection in more susceptible, aged (12-month-old) animals. SARS-MA15–infected animals rapidly lost weight and succumbed to infection (Supplementary Fig. 3a,b). SHC014-MA15 infection induced robust and sustained weight loss, but had minimal lethality. Trends in the histology and antigen staining patterns that we observed in young mice were conserved in the older animals (Supplementary Table 3). We excluded the possibility that SHC014-MA15 was mediating infection through an alternative receptor on the basis of experiments using Ace2−/− mice, which did not show weight loss or antigen staining after SHC014-MA15 infection (Supplementary Fig. 4a,b and Supplementary Table 2). Together, the data indicate that viruses with the SHC014 spike are capable of inducing weight loss in mice in the context of a virulent CoV backbone.
Given the preclinical efficacy of Ebola monoclonal antibody therapies, such as ZMApp10, we next sought to determine the efficacy of SARS-CoV monoclonal antibodies against infection with SHC014-MA15. Four broadly neutralizing human monoclonal antibodies targeting SARS-CoV spike protein had been previously reported and are probable reagents for immunotherapy11,12,13. We examined the effect of these antibodies on viral replication (expressed as percentage inhibition of viral replication) and found that whereas wild-type SARS-CoV Urbani was strongly neutralized by all four antibodies at relatively low antibody concentrations (Fig. 2a–d), neutralization varied for SHC014-MA15. Fm6, an antibody generated by phage display and escape mutants11,12, achieved only background levels of inhibition of SHC014-MA15 replication (Fig. 2a). Similarly, antibodies 230.15 and 227.14, which were derived from memory B cells of SARS-CoV–infected patients13, also failed to block SHC014-MA15 replication (Fig. 2b,c). For all three antibodies, differences between the SARS and SHC014 spike amino acid sequences corresponded to direct or adjacent residue changes found in SARS-CoV escape mutants (fm6 N479R; 230.15 L443V; 227.14 K390Q/E), which probably explains the absence of the antibodies’ neutralizing activity against SHC014. Finally, monoclonal antibody 109.8 was able to achieve 50% neutralization of SHC014-MA15, but only at high concentrations (10 μg/ml) (Fig. 2d). Together, the results demonstrate that broadly neutralizing antibodies against SARS-CoV may only have marginal efficacy against emergent SARS-like CoV strains such as SHC014.
SARS-CoV monoclonal antibodies have marginal efficacy against SARS-like CoVs.
(a–d) Neutralization assays evaluating efficacy (measured as reduction in the number of plaques) of a panel of monoclonal antibodies, which were all originally generated against epidemic SARS-CoV, against infection of Vero cells with SARS-CoV Urbani (black) or SHC014-MA15 (green). The antibodies tested were fm6 (n = 3 for Urbani; n = 5 for SHC014-MA15)11,12 (a), 230.15 (n = 3 for Urbani; n = 2 for SHC014-MA15) (b), 227.15 (n = 3 for Urbani; n = 5 for SHC014-MA15) (c) and 109.8 (n = 3 for Urbani; n = 2 for SHC014-MA15)13 (d). Each data point represents the group mean and error bars define the s.e.m. Note that the error bars in SARS-CoV Urbani–infected Vero cells in b,c are overlapped by the symbols and are not visible.
To evaluate the efficacy of existing vaccines against infection with SHC014-MA15, we vaccinated aged mice with double-inactivated whole SARS-CoV (DIV). Previous work showed that DIV could neutralize and protect young mice from challenge with a homologous virus14; however, the vaccine failed to protect aged animals in which augmented immune pathology was also observed, indicating the possibility of the animals being harmed because of the vaccination15. Here we found that DIV did not provide protection from challenge with SHC014-MA15 with regards to weight loss or viral titer (Supplementary Fig. 5a,b). Consistent with a previous report with other heterologous group 2b CoVs15, serum from DIV-vaccinated, aged mice also failed to neutralize SHC014-MA15 (Supplementary Fig. 5c). Notably, DIV vaccination resulted in robust immune pathology (Supplementary Table 4) and eosinophilia (Supplementary Fig. 5d–f). Together, these results confirm that the DIV vaccine would not be protective against infection with SHC014 and could possibly augment disease in the aged vaccinated group.
In contrast to vaccination of mice with DIV, the use of SHC014-MA15 as a live, attenuated vaccine showed potential cross-protection against challenge with SARS-CoV, but the results have important caveats. We infected young mice with 104 p.f.u. of SHC014-MA15 and observed them for 28 d. We then challenged the mice with SARS-MA15 at day 29 (Supplementary Fig. 6a). The prior infection of the mice with the high dose of SHC014-MA15 conferred protection against challenge with a lethal dose of SARS-MA15, although there was only a minimal SARS-CoV neutralization response from the antisera elicited 28 d after SHC014-MA15 infection (Supplementary Fig. 6b, 1:200). In the absence of a secondary antigen boost, 28 d.p.i. represents the expected peak of antibody titers and implies that there will be diminished protection against SARS-CoV over time16,17. Similar results showing protection against challenge with a lethal dose of SARS-CoV were observed in aged BALB/c mice with respect to weight loss and viral replication (Supplementary Fig. 6c,d). However, the SHC014-MA15 infection dose of 104 p.f.u. induced >10% weight loss and lethality in some aged animals (Fig. 1 and Supplementary Fig. 3). We found that vaccination with a lower dose of SHC014-MA15 (100 p.f.u.), did not induce weight loss, but it also failed to protect aged animals from a SARS-MA15 lethal dose challenge (Supplementary Fig. 6e,f). Together, the data suggest that SHC014-MA15 challenge may confer cross-protection against SARS-CoV through conserved epitopes, but the required dose induces pathogenesis and precludes use as an attenuated vaccine.
Having established that the SHC014 spike has the ability to mediate infection of human cells and cause disease in mice, we next synthesized a full-length SHC014-CoV infectious clone based on the approach used for SARS-CoV (Fig. 3a)2. Replication in Vero cells revealed no deficit for SHC014-CoV relative to that for SARS-CoV (Fig. 3b); however, SHC014-CoV was significantly (P < 0.01) attenuated in primary HAE cultures at both 24 and 48 h after infection (Fig. 3c). In vivo infection of mice demonstrated no significant weight loss but showed reduced viral replication in lungs of full-length SHC014-CoV infection, as compared to SARS-CoV Urbani (Fig. 3d,e). Together, the results establish the viability of full-length SHC014-CoV, but suggest that further adaptation is required for its replication to be equivalent to that of epidemic SARS-CoV in human respiratory cells and in mice.
Full-length SHC014-CoV replicates in human airways but lacks the virulence of epidemic SARS-CoV.
(a) Schematic of the SHC014-CoV molecular clone, which was synthesized as six contiguous cDNAs (designated SHC014A, SHC014B, SHC014C, SHC014D, SHC014E and SHC014F) flanked by unique BglI sites that allowed for directed assembly of the full-length cDNA expressing open reading frames (for 1a, 1b, spike, 3, envelope, matrix, 6–8 and nucleocapsid). Underlined nucleotides represent the overhang sequences formed after restriction enzyme cleavage. (b,c) Viral replication of SARS-CoV Urbani (black) or SHC014-CoV (green) after infection of Vero cells (b) or well-differentiated, primary air-liquid interface HAE cell cultures (c) at an MOI of 0.01. Samples were collected at individual time points with biological replicates (n = 3) for each group. Data represent one experiment for both Vero and HAE cells. (d,e) Weight loss (n = 3 for SARS-CoV MA15, n = 7 for SHC014-CoV; n = 6 for SARS-Urbani) (d) and viral replication in the lungs (n = 3 for SARS-Urbani and SHC014-CoV) (e) of 10-week-old BALB/c mice infected with 1 × 105 p.f.u. of SARS-CoV MA15 (gray), SHC014-CoV (green) or SARS-CoV Urbani (black) via the i.n. route. Each data point represents the group mean, and error bars define the s.e.m. **P < 0.01 and ***P < 0.001 using two-tailed Student’s t-test of individual time points.
During the SARS-CoV epidemic, links were quickly established between palm civets and the CoV strains that were detected in humans4. Building on this finding, the common emergence paradigm argues that epidemic SARS-CoV originated as a bat virus, jumped to civets and incorporated changes within the receptor-binding domain (RBD) to improve binding to civet Ace2 (ref. 18). Subsequent exposure to people in live-animal markets permitted human infection with the civet strain, which, in turn, adapted to become the epidemic strain (Fig. 4a). However, phylogenetic analysis suggests that early human SARS strains appear more closely related to bat strains than to civet strains18. Therefore, a second paradigm argues that direct bat-human transmission initiated SARS-CoV emergence and that palm civets served as a secondary host and reservoir for continued infection (Fig. 4b)19. For both paradigms, spike adaptation in a secondary host is seen as a necessity, with most mutations expected to occur within the RBD, thereby facilitating improved infection. Both theories imply that pools of bat CoVs are limited and that host-range mutations are both random and rare, reducing the likelihood of future emergence events in humans.
Coronavirus strains are maintained in quasi-species pools circulating in bat populations. (a,b) Traditional SARS-CoV emergence theories posit that host-range mutants (red circle) represent random and rare occurrences that permit infection of alternative hosts. The secondary-host paradigm (a) argues that a nonhuman host is infected by a bat progenitor virus and, through adaptation, facilitates transmission to humans; subsequent replication in humans leads to the epidemic viral strain. The direct paradigm (b) suggests that transmission occurs between bats and humans without the requirement of an intermediate host; selection then occurs in the human population with closely related viruses replicating in a secondary host, permitting continued viral persistence and adaptation in both. (c) The data from chimeric SARS-like viruses argue that the quasi-species pools maintain multiple viruses capable of infecting human cells without the need for mutations (red circles). Although adaptations in secondary or human hosts may be required for epidemic emergence, if SHC014 spike–containing viruses recombined with virulent CoV backbones (circles with green outlines), then epidemic disease may be the result in humans. Existing data support elements of all three paradigms.
Although our study does not invalidate the other emergence routes, it does argue for a third paradigm in which circulating bat CoV pools maintain ‘poised’ spike proteins that are capable of infecting humans without mutation or adaptation (Fig. 4c). This hypothesis is illustrated by the ability of a chimeric virus containing the SHC014 spike in a SARS-CoV backbone to cause robust infection in both human airway cultures and in mice without RBD adaptation. Coupled with the observation of previously identified pathogenic CoV backbones3,20, our results suggest that the starting materials required for SARS-like emergent strains are currently circulating in animal reservoirs. Notably, although full-length SHC014-CoV probably requires additional backbone adaption to mediate human disease, the documented high-frequency recombination events in CoV families underscores the possibility of future emergence and the need for further preparation.
To date, genomics screens of animal populations have primarily been used to identify novel viruses in outbreak settings21. The approach here extends these data sets to examine questions of viral emergence and therapeutic efficacy. We consider viruses with the SHC014 spike a potential threat owing to their ability to replicate in primary human airway cultures, the best available model for human disease. In addition, the observed pathogenesis in mice indicates a capacity for SHC014-containing viruses to cause disease in mammalian models, without RBD adaptation. Notably, differential tropism in the lung as compared to that with SARS-MA15 and attenuation of full-length SHC014-CoV in HAE cultures relative to SARS-CoV Urbani suggest that factors beyond ACE2 binding—including spike processivity, receptor bio-availability or antagonism of the host immune responses—may contribute to emergence. However, further testing in nonhuman primates is required to translate these finding into pathogenic potential in humans. Importantly, the failure of available therapeutics defines a critical need for further study and for the development of treatments. With this knowledge, surveillance programs, diagnostic reagents and effective treatments can be produced that are protective against the emergence of group 2b–specific CoVs, such as SHC014, and these can be applied to other CoV branches that maintain similarly heterogeneous pools.
In addition to offering preparation against future emerging viruses, this approach must be considered in the context of the US government–mandated pause on gain-of-function (GOF) studies22. On the basis of previous models of emergence (Fig. 4a,b), the creation of chimeric viruses such as SHC014-MA15 was not expected to increase pathogenicity. Although SHC014-MA15 is attenuated relative to its parental mouse-adapted SARS-CoV, similar studies examining the pathogenicity of CoVs with the wild-type Urbani spike within the MA15 backbone showed no weight loss in mice and reduced viral replication23. Thus, relative to the Urbani spike–MA15 CoV, SHC014-MA15 shows a gain in pathogenesis (Fig. 1). On the basis of these findings, scientific review panels may deem similar studies building chimeric viruses based on circulating strains too risky to pursue, as increased pathogenicity in mammalian models cannot be excluded. Coupled with restrictions on mouse-adapted strains and the development of monoclonal antibodies using escape mutants, research into CoV emergence and therapeutic efficacy may be severely limited moving forward. Together, these data and restrictions represent a crossroads of GOF research concerns; the potential to prepare for and mitigate future outbreaks must be weighed against the risk of creating more dangerous pathogens. In developing policies moving forward, it is important to consider the value of the data generated by these studies and whether these types of chimeric virus studies warrant further investigation versus the inherent risks involved.
Overall, our approach has used metagenomics data to identify a potential threat posed by the circulating bat SARS-like CoV SHC014. Because of the ability of chimeric SHC014 viruses to replicate in human airway cultures, cause pathogenesis in vivo and escape current therapeutics, there is a need for both surveillance and improved therapeutics against circulating SARS-like viruses. Our approach also unlocks the use of metagenomics data to predict viral emergence and to apply this knowledge in preparing to treat future emerging virus infections.
Viruses, cells, in vitro infection and plaque assays.
Wild-type SARS-CoV (Urbani), mouse-adapted SARS-CoV (MA15) and chimeric SARS-like CoVs were cultured on Vero E6 cells (obtained from United States Army Medical Research Institute of Infectious Diseases), grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, CA) and 5% fetal clone serum (FCS) (Hyclone, South Logan, UT) along with antibiotic/antimycotic (Gibco, Carlsbad, CA). DBT cells (Baric laboratory, source unknown) expressing ACE2 orthologs have been previously described for both human and civet; bat Ace2 sequence was based on that from Rhinolophus leschenaulti, and DBT cells expressing bat Ace2 were established as described previously8. Pseudotyping experiments were similar to those using an HIV-based pseudovirus, prepared as previously described10, and examined on HeLa cells (Wuhan Institute of Virology) that expressed ACE2 orthologs. HeLa cells were grown in minimal essential medium (MEM) (Gibco, CA) supplemented with 10% FCS (Gibco, CA) as previously described24. Growth curves in Vero E6, DBT, Calu-3 2B4 and primary human airway epithelial cells were performed as previously described8,25. None of the working cell line stocks were authenticated or tested for mycoplasma recently, although the original seed stocks used to create the working stocks are free from contamination. Human lungs for HAE cultures were procured under University of North Carolina at Chapel Hill Institutional Review Board–approved protocols. HAE cultures represent highly differentiated human airway epithelium containing ciliated and non-ciliated epithelial cells as well as goblet cells. The cultures are also grown on an air-liquid interface for several weeks before use, as previously described26. Briefly, cells were washed with PBS and inoculated with virus or mock-diluted in PBS for 40 min at 37 °C. After inoculation, cells were washed three times and fresh medium was added to signify time ‘0’. Three or more biological replicates were harvested at each described time point. No blinding was used in any sample collections nor were samples randomized. All virus cultivation was performed in a biosafety level (BSL) 3 laboratory with redundant fans in the biosafety cabinets, as described previously by our group2. All personnel wore powered air purifying respirators (Breathe Easy, 3M) with Tyvek suits, aprons and booties and were double-gloved.
Sequence clustering and structural modeling.
The full-length genomic sequences and the amino acid sequences of the S1 domains of the spike of representative CoVs were downloaded from Genbank or Pathosystems Resource Integration Center (PATRIC), aligned with ClustalX and phylogenetically compared by using maximum likelihood estimation using 100 bootstraps or by using the PhyML (https://code.google.com/p/phyml/) package, respectively. The tree was generated using maximum likelihood with the PhyML package. The scale bar represents nucleotide substitutions. Only nodes with bootstrap support above 70% are labeled. The tree shows that CoVs are divided into three distinct phylogenetic groups defined as α-CoVs, β-CoVs and γ-CoVs. Classical subgroup clusters are marked as 2a, 2b, 2c and 2d for β-CoVs, and 1a and 1b for the α-CoVs. Structural models were generated using Modeller (Max Planck Institute Bioinformatics Toolkit) to generate homology models for SHC014 and Rs3367 of the SARS RBD in complex with ACE2 based on crystal structure 2AJF (Protein Data Bank). Homology models were visualized and manipulated in MacPyMol (version 1.3).
Construction of SARS-like chimeric viruses.
Both wild-type and chimeric viruses were derived from either SARS-CoV Urbani or the corresponding mouse-adapted (SARS-CoV MA15) infectious clone (ic) as previously described27. Plasmids containing spike sequences for SHC014 were extracted by restriction digest and ligated into the E and F plasmid of the MA15 infectious clone. The clone was designed and purchased from Bio Basic as six contiguous cDNAs using published sequences flanked by unique class II restriction endonuclease sites (BglI). Thereafter, plasmids containing wild-type, chimeric SARS-CoV and SHC014-CoV genome fragments were amplified, excised, ligated and purified. In vitro transcription reactions were then preformed to synthesize full-length genomic RNA, which was transfected into Vero E6 cells as previously described2. The medium from transfected cells was harvested and served as seed stocks for subsequent experiments. Chimeric and full-length viruses were confirmed by sequence analysis before use in these studies. Synthetic construction of chimeric mutant and full-length SHC014-CoV was approved by the University of North Carolina Institutional Biosafety Committee and the Dual Use Research of Concern committee.
Ethics statement.
This study was carried out in accordance with the recommendations for the care and use of animals by the Office of Laboratory Animal Welfare (OLAW), NIH. The Institutional Animal Care and Use Committee (IACUC) of The University of North Carolina at Chapel Hill (UNC, Permit Number A-3410-01) approved the animal study protocol (IACUC #13-033) used in these studies.
Mice and in vivo infection.
Female, 10-week-old and 12-month-old BALB/cAnNHsD mice were ordered from Harlan Laboratories. Mouse infections were done as previously described20. Briefly, animals were brought into a BSL3 laboratory and allowed to acclimate for 1 week before infection. For infection and live-attenuated virus vaccination, mice were anesthetized with a mixture of ketamine and xylazine and infected intranasally, when challenged, with 50 μl of phosphate-buffered saline (PBS) or diluted virus with three or four mice per time point, per infection group per dose as described in the figure legends. For individual mice, notations for infection including failure to inhale the entire dose, bubbling of inoculum from the nose, or infection through the mouth may have led to exclusion of mouse data at the discretion of the researcher; post-infection, no other pre-established exclusion or inclusion criteria are defined. No blinding was used in any animal experiments, and animals were not randomized. For vaccination, young and aged mice were vaccinated by footpad injection with a 20-μl volume of either 0.2 μg of double-inactivated SARS-CoV vaccine with alum or mock PBS; mice were then boosted with the same regimen 22 d later and challenged 21 d thereafter. For all groups, as per protocol, animals were monitored daily for clinical signs of disease (hunching, ruffled fur and reduced activity) for the duration of the experiment. Weight loss was monitored daily for the first 7 d, after which weight monitoring continued until the animals recovered to their initial starting weight or displayed weight gain continuously for 3 d. All mice that lost greater than 20% of their starting body weight were ground-fed and further monitored multiple times per day as long as they were under the 20% cutoff. Mice that lost greater than 30% of their starting body weight were immediately sacrificed as per protocol. Any mouse deemed to be moribund or unlikely to recover was also humanely sacrificed at the discretion of the researcher. Euthanasia was performed using an isoflurane overdose and death was confirmed by cervical dislocation. All mouse studies were performed at the University of North Carolina (Animal Welfare Assurance #A3410-01) using protocols approved by the UNC Institutional Animal Care and Use Committee (IACUC).
Histological analysis.
The left lung was removed and submerged in 10% buffered formalin (Fisher) without inflation for 1 week. Tissues were embedded in paraffin and 5-μm sections were prepared by the UNC Lineberger Comprehensive Cancer Center histopathology core facility. To determine the extent of antigen staining, sections were stained for viral antigen using a commercially available polyclonal SARS-CoV anti-nucleocapsid antibody (Imgenex) and scored in a blinded manner by for staining of the airway and parenchyma as previously described20. Images were captured using an Olympus BX41 microscope with an Olympus DP71 camera.
Virus neutralization assays.
Plaque reduction neutralization titer assays were performed with previously characterized antibodies against SARS-CoV, as previously described11,12,13. Briefly, neutralizing antibodies or serum was serially diluted twofold and incubated with 100 p.f.u. of the different infectious clone SARS-CoV strains for 1 h at 37 °C. The virus and antibodies were then added to a 6-well plate with 5 × 105 Vero E6 cells/well with multiple replicates (n ≥ 2). After a 1-h incubation at 37 °C, cells were overlaid with 3 ml of 0.8% agarose in medium. Plates were incubated for 2 d at 37 °C, stained with neutral red for 3 h and plaques were counted. The percentage of plaque reduction was calculated as (1 − (no. of plaques with antibody/no. of plaques without antibody)) × 100.
Statistical analysis.
All experiments were conducted contrasting two experimental groups (either two viruses, or vaccinated and unvaccinated cohorts). Therefore, significant differences in viral titer and histology scoring were determined by a two-tailed Student’s t-test at individual time points. Data was normally distributed in each group being compared and had similar variance.
Biosafety and biosecurity.
Reported studies were initiated after the University of North Carolina Institutional Biosafety Committee approved the experimental protocol (Project Title: Generating infectious clones of bat SARS-like CoVs; Lab Safety Plan ID: 20145741; Schedule G ID: 12279). These studies were initiated before the US Government Deliberative Process Research Funding Pause on Selected Gain-of-Function Research Involving Influenza, MERS and SARS Viruses (http://www.phe.gov/s3/dualuse/Documents/gain-of-function.pdf). This paper has been reviewed by the funding agency, the NIH. Continuation of these studies was requested, and this has been approved by the NIH.
SARS-CoV is a select agent. All work for these studies was performed with approved standard operating procedures (SOPs) and safety conditions for SARS-CoV, MERs-CoV and other related CoVs. Our institutional CoV BSL3 facilities have been designed to conform to the safety requirements that are recommended in the Biosafety in Microbiological and Biomedical Laboratories (BMBL), the US Department of Health and Human Services, the Public Health Service, the Centers for Disease Control (CDC) and the NIH. Laboratory safety plans were submitted to, and the facility has been approved for use by, the UNC Department of Environmental Health and Safety (EHS) and the CDC. Electronic card access is required for entry into the facility. All workers have been trained by EHS to safely use powered air purifying respirators (PAPRs), and appropriate work habits in a BSL3 facility and active medical surveillance plans are in place. Our CoV BSL3 facilities contain redundant fans, emergency power to fans and biological safety cabinets and freezers, and our facilities can accommodate SealSafe mouse racks. Materials classified as BSL3 agents consist of SARS-CoV, bat CoV precursor strains, MERS-CoV and mutants derived from these pathogens. Within the BSL3 facilities, experimentation with infectious virus is performed in a certified Class II Biosafety Cabinet (BSC). All members of the staff wear scrubs, Tyvek suits and aprons, PAPRs and shoe covers, and their hands are double-gloved. BSL3 users are subject to a medical surveillance plan monitored by the University Employee Occupational Health Clinic (UEOHC), which includes a yearly physical, annual influenza vaccination and mandatory reporting of any symptoms associated with CoV infection during periods when working in the BSL3. All BSL3 users are trained in exposure management and reporting protocols, are prepared to self-quarantine and have been trained for safe delivery to a local infectious disease management department in an emergency situation. All potential exposure events are reported and investigated by EHS and UEOHC, with reports filed to both the CDC and the NIH.
Research in this manuscript was supported by grants from the National Institute of Allergy & Infectious Disease and the National Institute of Aging of the US National Institutes of Health (NIH) under awards U19AI109761 (R.S.B.), U19AI107810 (R.S.B.), AI085524 (W.A.M.), F32AI102561 (V.D.M.) and K99AG049092 (V.D.M.), and by the National Natural Science Foundation of China awards 81290341 (Z.-L.S.) and 31470260 (X.-Y.G.), and by USAID-EPT-PREDICT funding from EcoHealth Alliance (Z.-L.S.). Human airway epithelial cultures were supported by the National Institute of Diabetes and Digestive and Kidney Disease of the NIH under award NIH DK065988 (S.H.R.). We also thank M.T. Ferris (Dept. of Genetics, University of North Carolina) for the reviewing of statistical approaches and C.T. Tseng (Dept. of Microbiology and Immunology, University of Texas Medical Branch) for providing Calu-3 cells. Experiments with the full-length and chimeric SHC014 recombinant viruses were initiated and performed before the GOF research funding pause and have since been reviewed and approved for continued study by the NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
V.D.M. designed, coordinated and performed experiments, completed analysis and wrote the manuscript. B.L.Y. designed the infectious clone and recovered chimeric viruses; S.A. completed neutralization assays; L.E.G. helped perform mouse experiments; T.S. and J.A.P. completed mouse experiments and plaque assays; X.-Y.G. performed pseudotyping experiments; K.D. generated structural figures and predictions; E.F.D. generated phylogenetic analysis; R.L.G. completed RNA analysis; S.H.R. provided primary HAE cultures; A.L. and W.A.M. provided critical monoclonal antibody reagents; and Z.-L.S. provided SHC014 spike sequences and plasmids. R.S.B. designed experiments and wrote manuscript.
1. Ge XY, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
2. Yount B, et al. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2003;100:12995–13000. doi: 10.1073/pnas.1735582100. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
3. Becker MM, et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA. 2008;105:19944–19949. doi: 10.1073/pnas.0808116105. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
6. He B, et al. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome–like coronavirus from bats in China. J. Virol. 2014;88:7070–7082. doi: 10.1128/JVI.00631-14. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
7. Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Res. 2013;100:246–254. doi: 10.1016/j.antiviral.2013.08.014. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
8. Sheahan T, et al. Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. J. Virol. 2008;82:2274–2285. doi: 10.1128/JVI.02041-07. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Yoshikawa T, et al. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome–associated coronavirus infection. PLoS ONE. 2010;5:e8729. doi: 10.1371/journal.pone.0008729. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
10. Qiu X, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514:47–53. doi: 10.1038/nature13777. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
11. Sui J, et al. Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog. 2008;4:e1000197. doi: 10.1371/journal.ppat.1000197. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
12. Sui J, et al. Effects of human anti–spike protein receptor binding domain antibodies on severe acute respiratory syndrome coronavirus neutralization escape and fitness. J. Virol. 2014;88:13769–13780. doi: 10.1128/JVI.02232-14. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
13. Rockx B, et al. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2010;201:946–955. doi: 10.1086/651022. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
14. Spruth M, et al. A double-inactivated whole-virus candidate SARS coronavirus vaccine stimulates neutralizing and protective antibody responses. Vaccine. 2006;24:652–661. doi: 10.1016/j.vaccine.2005.08.055. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
15. Bolles M, et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 2011;85:12201–12215. doi: 10.1128/JVI.06048-11. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
17. Deming D, et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 2006;3:e525. doi: 10.1371/journal.pmed.0030525. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
18. Graham RL, Donaldson EF, Baric RS. A decade after SARS: strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013;11:836–848. doi: 10.1038/nrmicro3143. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
19. Graham RL, Baric RS. Recombination, reservoirs and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 2010;84:3134–3146. doi: 10.1128/JVI.01394-09. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Agnihothram S, et al. A mouse model for betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. MBio. 2014;5:e00047–14. doi: 10.1128/mBio.00047-14. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
21. Relman DA. Metagenomics, infectious disease diagnostics and outbreak investigations: sequence first, ask questions later? J. Am. Med. Assoc. 2013;309:1531–1532. doi: 10.1001/jama.2013.3678. [PubMed] [CrossRef] [Google Scholar]
23. Frieman M, et al. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J. Virol. 2012;86:884–897. doi: 10.1128/JVI.05957-11. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
24. Ren W, et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 2008;82:1899–1907. doi: 10.1128/JVI.01085-07. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
25. Sims AC, et al. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J. Virol. 2013;87:3885–3902. doi: 10.1128/JVI.02520-12. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
26. Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005;107:183–206. [PubMed] [Google Scholar]
27. Roberts Anjeanette, Deming Damon, Paddock Christopher D., Cheng Aaron, Yount Boyd, Vogel Leatrice, Herman Brian D., Sheahan Tim, Heise Mark, Genrich Gillian L., Zaki Sherif R., Baric Ralph, Subbarao Kanta. A Mouse-Adapted SARS-Coronavirus Causes Disease and Mortality in BALB/c Mice. PLoS Pathogens. 2007;3(1):e5. doi: 10.1371/journal.ppat.0030005. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Almamlouk R, Kashour T, Obeidat S, Bois MC, Maleszewski JJ, Omrani OA, Tleyjeh R, Berbari E, Chakhachiro Z, Zein-Sabatto B, Gerberi D, Tleyjeh IM; Cardiac Autopsy in COVID-19 Study Group; Paniz Mondolfi AE, Finn AV, Duarte-Neto AN, Rapkiewicz AV, Frustaci A, Keresztesi AA, Hanley B, Märkl B, Lardi C, Bryce C, Lindner D, Aguiar D, Westermann D, Stroberg E, Duval EJ, Youd E, Bulfamante GP, Salmon I, Auer J, Maleszewski JJ, Hirschbühl K, Absil L, Barton LM, Ferraz da Silva LF, Moore L, Dolhnikoff M, Lammens M, Bois MC, Osborn M, Remmelink M, Nascimento Saldiva PH, Jorens PG, Craver R, Aparecida de Almeida Monteiro R, Scendoni R, Mukhopadhyay S, Suzuki T, Mauad T, Fracasso T, Grimes Z. COVID-19-Associated cardiac pathology at the postmortem evaluation: a collaborative systematic review. Clin Microbiol Infect. 2022 Aug;28(8):1066-1075. doi: 10.1016/j.cmi.2022.03.021. Epub 2022 Mar 23. PMID: 35339672; PMCID: PMC8941843.
Abstract
Background
Many postmortem studies address the cardiovascular effects of COVID-19 and provide valuable information, but are limited by their small sample size.
Objectives
The aim of this systematic review is to better understand the various aspects of the cardiovascular complications of COVID-19 by pooling data from a large number of autopsy studies.
Data sources
We searched the online databases Ovid EBM Reviews, Ovid Embase, Ovid Medline, Scopus, and Web of Science for concepts of autopsy or histopathology combined with COVID-19, published between database inception and February 2021. We also searched for unpublished manuscripts using the medRxiv services operated by Cold Spring Harbor Laboratory.
Study eligibility criteria
Articles were considered eligible for inclusion if they reported human postmortem cardiovascular findings among individuals with a confirmed SARS coronavirus type 2 (CoV-2) infection.
Participants
Confirmed COVID-19 patients with post-mortem cardiovascular findings.
Interventions
None.
Methods
Studies were individually assessed for risk of selection, detection, and reporting biases. The median prevalence of different autopsy findings with associated interquartile ranges (IQRs).
Results
This review cohort contained 50 studies including 548 hearts. The median age of the deceased was 69 years. The most prevalent acute cardiovascular findings were myocardial necrosis (median: 100.0%; IQR, 20%–100%; number of studies = 9; number of patients = 64) and myocardial oedema (median: 55.5%; IQR, 19.5%–92.5%; number of studies = 4; number of patients = 46). The median reported prevalence of extensive, focal active, and multifocal myocarditis were all 0.0%. The most prevalent chronic changes were myocyte hypertrophy (median: 69.0%; IQR, 46.8%–92.1%) and fibrosis (median: 35.0%; IQR, 35.0%–90.5%). SARS-CoV-2 was detected in the myocardium with median prevalence of 60.8% (IQR 40.4-95.6%).
Conclusions
Our systematic review confirmed the high prevalence of acute and chronic cardiac pathologies in COVID-19 and SARS-CoV-2 cardiac tropism, as well as the low prevalence of myocarditis in COVID-19.
Preexisting cardiovascular comorbidities are prevalent among patients with COVID-19 and associated with a higher mortality rate [[1], [2], [3]]. For example, in the study reported by the Chinese Centre for Disease Control and Prevention describing the early experience with the epidemic in the Hubie province, patients with cardiovascular comorbidities had a case fatality rate of 10.5% compared with an overall cohort fatality rate of 2.3% [4]. There is also emerging robust evidence to suggest long-term cardiovascular sequalae after acute COVID-19 infection with an increased risk of incident conditions, including dysrhythmias, ischemic and nonischemic heart disease, myocarditis, and thromboembolic disease, among different COVID-19 disease severity groups compared with patients not infected with COVID-19 [5].
In addition, echocardiographic studies in populations infected with COVID-19 have demonstrated a high prevalence of ventricular dysfunction. In a prospective international study of 1216 patients with COVID-19, overall left and right ventricular dysfunction were reported in 39% and 33%, respectively [6]. Even in patients without preexisting cardiac disease, abnormal echocardiographic findings were evident in 46% of patients, with 13% manifesting severe abnormalities [6]. Acute myocardial injury manifesting as an elevation in cardiac troponins has been reported in 7% to 28% of patients with COVID-19 [[7], [8], [9], [10]]. Such acute cardiac injury was associated with higher overall mortality [10]. In a meta-analysis of 13 studies, the risk of death was high among patients with COVID-19 who had acute myocardial injury as defined by elevated serum troponins (risk ratio: 7.95; CI, 5.12–12.34; p <0.001; I2 = 65%) [11].
Several mechanisms have been proposed to explain acute myocardial injury and ventricular dysfunction in patients with COVID-19, including supply–demand mismatch secondary to hypoxemia and elevated cardiac demand, direct damage inflicted by inflammatory cytokines, microvascular dysfunction, myocarditis, coagulation abnormalities, and coronary artery plaque instability [12,13]. Other proposed mechanisms, such as vasospasm, microvascular thrombosis, and myocarditis, could be responsible for the ST-segment elevation [14].
A direct pathologic cardiovascular examination of decedents provides important information about the true frequency of cardiac complications among patients with COVID-19, and sheds light on possible pathologic mechanisms. Early on, small postmortem studies described evidence of myocardial inflammation associated with myocyte necrosis in patients with COVD-19 [15,16], as well as a possible direct SARS coronavirus type 2 (CoV-2) infection of the heart [17]. Moreover, nonspecific longstanding findings, such as cardiac hypertrophy and fibrosis, suggest underlying cardiovascular disease in a subset of these patients. Multiple subsequent studies have been published with varying sample sizes, methodologies, and findings. These studies provide valuable information about the nature of cardiac involvement in patients with COVID-19. However, their small sample sizes make deriving a clear picture of the true frequencies of cardiovascular complications in this novel disease challenging. In this international collaboration, we undertook a systematic review to better understand the pathologic cardiac findings in patients with COVID-19 at the time of postmortem evaluation.
We followed the Preferred Reporting Items for Systematic Reviews and Meta-analyses guidelines. The protocol of this review was registered in PROSPERO (CRD42020223551).
Literature search and study selection
The literature was searched by a medical librarian for the concepts of autopsy or histopathology combined with COVID-19. The search strategies were created using keywords and standardized index terms (Doc. S1). Searches were run in February 2021 in Ovid EBM Reviews, Ovid Embase (1974+), Ovid Medline (1946+, including ePUB ahead of print, in-process, and other nonindexed citations), Scopus (1970+), and Web of Science (1975+). We also searched for unpublished manuscripts using the medRxiv services operated by Cold Spring Harbor Laboratory. In addition, we searched Google Scholar and the references of eligible studies and review articles.
Articles were considered eligible for inclusion if they were studies with human participants and reported cardiac autopsy findings among individuals with a SARS-CoV-2 infection. We included studies published in any language.
Identification of studies
Two reviewers (RA and SO) examined the titles and abstracts of articles using the studies selection criteria. Then, they examined the full texts to confirm that each article met the eligibility criteria.
Data collection
Data were extracted by two reviewers (R.A. and S.O.) and in duplicates into a prespecified data collection form. Disagreements were discussed with the senior reviewers (I.T. and T.K.). Data were collected on the following prespecified outcomes: 1) Study location, study type, number of cases, patient selection, selection bias, and autopsy type; 2) baseline characteristics, including age, sex, ethnicity, body mass index, cause of death, days to death, and presence of comorbidities; 3) laboratory test values, including maximum serum troponin levels, serum brain natriuretic peptide, serum ferritin, and D-dimer levels; 4) cardiac autopsy findings; and 5) ultrastructural studies, including immunohistochemistry and electron microscopy. The Cardiac Autopsy in COVID-19 Study Group collaborators completed a data collection form (Doc. S2).
One author assessed the studies for risk of selection, detection, and reporting biases. Specifically, studies were evaluated on whether consecutively deceased patients with COVID-19 underwent a cardiac autopsy to reduce selection bias.
Statistical analyses
The number and percentage of patients manifesting different findings during cardiac autopsies were extracted from each study and confirmed with the studies’ authors. We initially planned to perform meta-analyses to obtain pooled estimates of the different findings’ prevalences. However, this was not possible due to the limited number of studies that performed consecutive cardiac autopsies. We report the median prevalence of cardiac autopsy findings across studies with sample sizes ≥5, with associated interquartile ranges (IQRs) (see Table 1 ).
Table 1
Summary of median prevalence of cardiac autopsy findings of studies with ≥5 patients
The search yielded 4760 results. We examined the entire text of 58 manuscripts after removing duplicates and screening the titles. However, eight studies were excluded, leaving 50 studies with 548 hearts in the final cohort (Fig. 1 ). Most studies were case reports (n = 13) or case series (n = 37; Doc. S3). Autopsy cases were acquired from manuscripts spanning experiences from 15 countries. The number of cases per study ranged from 1 to 80 (median: 4.5), and five cases were identified as reporting consecutive autopsies (encompassing 155 subjects) [[18], [19], [20], [21], [22]]. There were 42 minimally invasive autopsies, 102 partial autopsies, and 301 complete autopsies among the autopsies where completeness was stated or could be inferred.
Preferred Reporting Items for Systematic Reviews and Meta-analyses 2009 flow diagram.
Patient demographics, comorbidities, and cause of death
The median age of the deceased was 69 years (range, 22–97 years; n = 548), and 62% of cases were men (n = 338 of 548). The most common comorbidities were systemic hypertension (n = 298; 56%) and coronary artery disease (n = 252; 49%). Other less common comorbidities included chronic obstructive pulmonary disease, diabetes, obesity, chronic kidney disease, old myocardial infarction, dementia, malignancy, and sleep apnoea (Fig. 2 ). Elevated troponin was demonstrated in 55% of cases.
Bar chart showing reported comorbidities of deceased patients included in this cohort. CAD, coronary artery disease; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; MI, myocardial infarction. Data labels show the prevalence of reported comorbidities (can overlap in a single patient).
The cause of death was reported in 479 cases, with the most reported being respiratory in origin. However, in 62 cases, cardiac involvement was identified as a key factor in mortality. The median time from the onset of symptoms to death was 9 days (range, 0–71 days; n = 401).
Cardiovascular autopsy findings
General findings
Cardiac abnormalities were found in gross pathology or histology test results in almost all cases. Heart weights were available for 276 hearts (51%), with a median weight of 465 g (range, 238–1070 g).
SARS coronavirus type to infection of the heart
Nineteen studies [[17], [18], [19], [23], [24], [25], [26], [27], [28]] with 217 cases explored the presence and localization of SARS-CoV-2 infection in the heart using different modalities, including RT-PCR, immunohistochemistry, in situ hybridization, and electron microscopy. Ten studies [18,19,[26], [27], [28],30,32,33,35,37] with a total of 116 cases detected SARS-CoV-2 infection in the cardiac tissues in 70 cases with a median of 60.8% (IQR, 40.4%–95.6%; Fig. 3 ).
Box-and-whisker plot of cardiac autopsy findings of studies with ≥5 patients as median percentage prevalence and associated interquartile ranges. CAD, coronary artery disease.
Active replication of SARS-CoV-2 within the heart was determined using the RNA scope in situ hybridization technique looking for the presence of the negative strand of the SARS-CoV-2 viral RNA or through the identification of subgenomic RNA, both of which indicate active viral replication. Four investigators employed these techniques in 55 cases [18,27,28,33], and verified the presence of active SARS-CoV-2 viral replication in 15 hearts (27%).
Localization of SARS-CoV-2 within different cardiac cell compartments was studied by nine investigators [23,25,26,30,33,34,[36], [37], [38]] in 56 hearts from total of 95 cases using electron microscopy or immunohistochemistry. The presence of SARS-CoV-2 infection within the cardiomyocytes was reported in 11 hearts by four investigators [30,33,34,36]. SARS-CoV-2 infection was also detected in cardiac vascular endothelial cells in seven hearts and in cardiac fibroblasts in one heart [26,34]. On the other hand, other investigators [23,25,26,37,38] could not detect SARS-CoV-2 infection within any cell type in the heart.
Myocarditis
The majority of studies did not specify what definition of myocarditis was used. However, we inferred from the description of the histopathological findings that the Dallas criteria were used by most studies. Several investigators used immunohistochemical studies with different antibodies to identify subtypes of cellular infiltrates, but most did not use immunohistochemical criteria to diagnose myocarditis. In total, 36 cases had myocarditis and 16 had inflammatory infiltrates but no myocyte damage (Fig. 3).
Few cases reported extensive myocarditis, ranging from 0.0% to 19.3%, with a median of 0.0% across 10 studies [20,23,26,28,32,37,[39], [40], [41], [42]] with a total of 175 cases (Fig. 3). Grosse et al., who authored the only consecutive study to report a prevalence for this finding, did not find any cases of extensive myocarditis across 14 cases. Focal active myocarditis was reported by 13 studies [20,21,23,25,26,28,32,[37], [38], [39], [40],42,43], ranging from 0.0% to 55.5%. Nine studies [19,20,26,28,32,37,39,42,44] with total of 131 cases described multifocal myocarditis with a median prevalence of 0.0% (IQR, 0.0%–2.1%). Finally, 15 studies [18,[20], [21], [22],[26], [27], [28],32,35,37,39,40,42,44,45] with 279 cases reported infiltrates without myocyte damage with a median prevalence of 0.6% (range, 0.0%–28.9%; Fig. 3).
Other acute cardiac pathologic changes
Necrosis had the highest median reported prevalence across nine studies, of which none were considered consecutive studies, including 64 autopsies [28,29,31,34,40,43,[46], [47], [48]] with a median of 100% (n = 64; IQR, 20.0%–100%). This was followed by cardiac interstitial oedema (n = 46; median: 55%; IQR, 19.5%–92.5%) [22,27,30,42], with Duarte-Neto et al. reporting a prevalence of 90% across ten consecutive autopsies (Fig. 3).
Microvessel thrombi had the highest reported median prevalence across the category of thromboembolic disease among eight studies, reporting a similar prevalence across the studies [22,23,27,28,30,38,44,49] with 43 of 103 cases (median: 36.2%; IQR, 17.5%–61.7%). Alternatively, acute myocardial infarction had the lowest reported median prevalence in this category (median: 11.8%; IQR, 7.9%–13.8%; Fig. 3). Acute epi-pericarditis was reported with a median prevalence of 15.5% (IQR, 11.9%–19.2%) across six studies [28,30,33,38,39,41] in 29 of 110 cases, and small vessel vasculitis had a median reported prevalence of 28.6% (IQR, 16.0%–32.5%) across three studies [38,41,43] in 12 of 86 cases (Fig. 3). Other less frequently reported findings include single-cell ischemia in one of seven patients [35], myocyte ischemic degeneration with pyknosis in one case report [50], and contraction bands in one of three cases [51].
Chronic cardiac findings
Hypertrophy was the most common pathological finding with a median of 69.0% (IQR, 46.8%–92.1%) across 18 studies [[18], [19], [20],22,23,25,28,[31], [32], [33],35,38,39,[41], [42], [43],45,49] in 197 of 303 cases. Fibrosis was reported in 13 studies [20,22,25,27,28,[30], [31], [32],37,[42], [43], [44], [45]] with a median of 42.9% (IQR, 35.0%–90.5%) in 104 of 183 cases. Among these 13 studies, ten studies reported various details about the nature of fibrotic changes [20,25,27,28,30,31,37,42,44,45]. Two studies [42,44] reported on the severity of fibrosis with 32 cases (20 with mild and 8 with moderate fibrosis). Six studies [20,27,28,30,31,44] reported on the extent of fibrosis in a total of 98 cases. There was fibrosis in 46 of these cases, which was diffuse in 18 cases and focal or patchy in 28 cases. Replacement fibrosis was described by two authors [25,27] in eight cases. Eight studies [18,20,23,25,28,31,35,45,49] with 131 patients observed amyloidosis in 21 cases with a median prevalence of 13.6% (IQR, 9.8%–17.4%). The type of amyloidosis was reported in 14 of these cases, and determined to be transthyretin in 13 cases and amyloid P in one case, with patient age ranging between 71 and 96 years [20,23,28,31,35,45]. Other less-reported pathologies were chronic pericarditis, reported by three studies in 17 of 47 cases [28,33,43], and ischemic heart disease in 1 of 3 cases in one study [50].
This systematic review of pathology-derived cardiac changes in patients with COVID-19 included 50 studies with more than 500 cases, and was the effort of an international collaboration. The most prevalent chronic changes were myocardial hypertrophy, underlying coronary artery disease, and fibrosis (median: 69.0%, 46.2%, and 42.9%, respectively). The high prevalence of chronic cardiac pathologies among patients who died due to COVID-19 supports the findings from previously published epidemiologic studies [[1], [2], [3]].
Interestingly, another underlying cardiac disease, amyloidosis, was reported in a median of 13.6% of patients with COVID-19, with patient age ranging between 71 and 96 years. The overall prevalence of cardiac amyloidosis in an unselected, sequential autopsy population was reported at approximately 4% [23]. Conversely, a Finnish autopsy study of individuals age >85 years detected cardiac amyloidosis in 25% of cases [52]. Although cardiac amyloidosis prevalence almost certainly increases with patient age, cardiac amyloidosis is also likely underdiagnosed, particularly among patients with heart failure and preserved ejection fraction [[53], [54], [55]]. Nevertheless, the relatively high proportion of cardiac amyloidosis among decedents with COVID-19 compared with unselected autopsy rates suggests that this condition may render patients vulnerable to adverse outcomes from SARS-CoV-2 infection. This is further supported by the average age of patients with cardiac amyloidosis who died of COVID-19. Possible mechanisms for this complication have been proposed; however, decreased cardiac reserve innate to underlying cardiovascular disease, including amyloidosis, likely plays a significant role [56].
The prevalence of acute thromboembolic pathologies in descending frequency included microvessel thrombi (36.2%), pulmonary embolism (22.2%), cardiac large vessel thrombosis (14.3%), and acute myocardial infarction (11.8%). The increased cardiac and pulmonary vascular thrombi correlate strongly with the clinical evidence of increased thromboembolic phenomena in patients with COVID-19. Moreover, these thrombotic changes, along with the observed high prevalence of acute cardiac injuries (e.g. necrosis, oedema, and epi-pericarditis) are concordant with the clinically documented ventricular dysfunction [6] and serologic markers of cardiac injury, such as increased troponins [[7], [8], [9], [10]]. In fact, microvascular thrombosis has been cited as the causative agent of cardiac injury in most decedents with COVID-19 [57]. This finding dovetails with the lower prevalence of large vessel cardiac thrombosis, and is consistent with the coronary angiographic findings in patients with COVID-19, wherein a culprit lesion was not identified in more than 40% of patients with suspected acute myocardial infarction [14].
SARS-CoV-2 cardiac tropism
SARS-CoV-2 gains entry into the host cells through the binding of its spike protein to the angiotensin-converting enzyme 2 with the help of the host transmembrane protease serine 2 [58]. Both proteins have been shown to be expressed in the heart [23,[59], [60], [61]]. The predecessor of SARS-CoV-2 (SARS-CoV and its associated syndrome SARS) also uses the angiotensin-converting enzyme 2 protein for cell entry, and has been shown to infect the heart and induce inflammatory changes based on data from the first decade of the 21st century [61]. These findings, along with the clinical observations of acute cardiac injury among patients with COVID-19, prompted several investigators to address three important questions: 1) Is SARS-CoV-2 present in the hearts of decedents with COVID-19; 2) if so, which cell type(s) does SARS-CoV-2 infect; and 3) can SARS-CoV-2 replicate in heart tissues?
Twenty studies explored the presence of SARS-CoV-2 within the heart, the majority of which targeted the identification of SARS-CoV-2 RNA in heart tissues using RT-PCR or in situ hybridization. Other employed techniques included immunohistochemistry to identify SARS-CoV-2 proteins (e.g. spike or nucleocapsid protein), as well as electron microscopy. These investigators identified the presence of SARS-CoV-2 in almost 60% of the examined hearts. Additionally, a few investigators identified SARS-CoV-2 replication within heart tissues in several cases [18,27,28,33]. Furthermore, studies investigated cell-type localization of SARS-CoV-2 within the heart, and provided evidence of the presence of SARS-CoV-2 viral particles within the cardiomyocytes [30,33,34,36]. Bulfamante et al. observed degenerative changes in cardiomyocytes containing SARS-CoV-2 viral particles [33]. These findings are supported by a study that demonstrated SARS-CoV-2 infection and propagation in induced pluripotent stem cell-derived cardiomyocytes [62]. SARS-CoV-2 has also been found in vascular endothelial cells and cardiac fibroblasts [26,34]. These reports establish SARS-CoV-2 cardiac tropism, and present a possible link between SARS-CoV-2 and certain acute cardiac pathologies (e.g. myocarditis). However, although RT-PCR represents a time-efficient method to determine tissue positive for SARS-CoV-2, RT-PCR does not allow for tissue localization. Wong et al. suggested and attempted to validate a Fluorescence In Situ Hybridization (FISH) method using positive and negative controls by detecting endogenous human genes (POLR2A and PPIB) and a bacterial gene (dap gene of Bacillus subtilis) to allow for a tissue-specific analysis [63].
SARS-CoV-2-induced myocarditis
In this review, we subdivided the reported cardiac inflammatory processes in patients with COVID-19 into four categories based on the degree of myocardial involvement and the presence of associated myocyte damage. Overall, the prevalence of each category was low, with vast differences between individual studies that cannot be explained solely by the methodological differences of the studies, and likely indicate significant selection and reporting bias. The median reported prevalence of extensive myocarditis, multifocal active myocarditis, and focal active myocarditis were all 0.0%, and the median prevalence of inflammatory infiltrate without myocyte damage was 0.6%.
Regrettably, clinical correlation or pooled prevalence estimates in the included reported autopsy series were not possible due to the heterogenous results and paucity of clinical and imaging data provided. Nonetheless, reports of clinically diagnosed myocarditis with pathologic correlation have been reported among inpatients with COVID-19 [36,48,64]. Intriguingly, Gauchotte et al. demonstrated pathologic evidence of myocarditis without lung involvement, and further showed the presence of the SARS-CoV-2 genome in cardiomyocytes in this case [36]. This finding is concordant with other studies suggesting a greater degree of inflammation with viral presence in the heart [65].
The diagnosis of most cases of myocarditis included in this review were based on the Dallas criteria. This methodology, although widely accepted, is not without its inherent limitations. First and foremost, the Dallas criteria were developed to diagnose myocarditis by endomyocardial biopsy (EMB), not autopsy, wherein a more abundant amount of tissue is available for histologic evaluation. The generalization (and clinical significance) of small foci of myocyte damage within autopsy-derived cardiac tissue is challenging to ascertain. Other limitations of the Dallas criteria include significant interobserver variability and sampling errors [66,67].
Although less of an issue in autopsy-derived tissue, the focal nature of the disease leads to sampling errors that have been shown to compromise the sensitivity of the histopathological diagnosis of myocarditis by EMB [68,69]. Chow et al. had estimated that a mean of 17 samples per patient would be required to establish a diagnosis of myocarditis [69], which likely explains why examining an increased number of cardiac tissue blocks at the time of autopsy resulted in a greater likelihood of identifying focal myocarditis.
The overall low prevalence of myocarditis in patients with COVID-19 is of interest, particularly when placed in the greater context of the available literature. In a recent meta-analysis on the diagnosis of myocarditis by EMB (including 61 studies with 10,491 patients), the prevalence of myocarditis according to the Dallas criteria was 8.04% [70]. This diagnosis was made on the relatively limited amount of tissue provided by EMB. In contrast, this review shows a myocarditis prevalence of 8% in abundant available tissue, often comprising multiple blocks of myocardium with greater orders of magnitude in the amount of tissue to examine. The pretest factors among these data points differ, but underscores the overall low prevalence of myocarditis in COVID-19 deaths and is concordant with previous literature reviews on the topic [71].
Prior studies have shown the added sensitivity of immunohistochemistry in the diagnosis of myocarditis. Katzmann et al. showed that the sensitivity of the Dallas criteria in detecting myocarditis was much lower than when immunohistochemistry is utilized, with a detection rate of 50.8% (vs. 8.04% without immunohistochemistry) [70]. In another study of 84 cases of myocarditis based on the immunohistochemistry criteria, applying the Dallas criteria without immunohistochemistry would have categorized only 8% of these case as active myocarditis [72].
The true prevalence of myocarditis in COVID-19 remains very hard to determine from the current autopsy and imaging studies, the latter of which shows a discordantly high prevalence of myocarditis compared with postmortem examinations. A recent systematic review of cardiovascular magnetic resonance findings in COVID-19 including 199 patients showed that myocarditis was the most prevalent diagnosis (40.2%) [73]. Future studies should integrate clinical imaging and more rigorous and systematic autopsy studies to help resolve this issue. Such initiative should be conducted in the form of an international registry that uses a unified autopsy examination and imaging protocols in accordance with published guidelines [74,75].
Limitations
This systematic review included the largest number of studies and cases published to date on cardiac changes in fatal COVID-19 with both qualitative and quantitative analyses of different cardiac pathologies observed in COVID-19. However, our study has several limitations. First, the majority of the included studies were small. Second, these studies were heterogeneous in their methodologies and patient cultural origins, with very few studies performing consecutive autopsies, which makes meta-analyses unfeasible. Moreover, selection and reporting bias likely affected most included studies, as evidenced by the nonconsecutive nature of case recruitment and the very high differences between the studies in the perveances of reported pathologies.
Our systematic review confirmed the high prevalence of acute and chronic cardiac pathologies in the autopsy-derived hearts of decedents with COVID-19. These findings help explain observations from clinical epidemiologic studies, such as thromboembolic phenomena and acute myocardial injury. Our study also provides evidence for SARS-CoV-2 cardiac tropism, and confirmed the low prevalence of myocarditis in patients with COVID-19.
IMT designed the study. IMT, RA, SO, and MCB coordinated the study. DG designed and ran the literature search. RA, SO, OAO, RT, ZC, BZS, EB, and TK acquired the data, screened records, and extracted the data. IMT and OAO conducted the formal analyses. TK wrote the report with input from MCB and JJM. All authors provided critical conceptual input, analyzed and interpreted data, and critically revised the report.
Elie Berbari reports royalties or licenses from UTD of <$5000 per year. Amy V. Rapkiewicz reports payment for expert testimony by Eric Hack, Esq. Bruno Märkl reports grants or contracts from the German Registry of COVID-19 Autopsies, funded by the Federal Ministry of Health and the Federal Ministry of Education and Research within the framework of the network of university medicine. Diana Lindner reports support for the present manuscript, grants, and contracts from the German Centre for Cardiovascular Research, Deutsche Herzstiftung. Dirk Westermann reports consulting fees and payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Abiomed, Bayer, AstraZeneca, Novartis, and Medtronic. Klaus Hirschbühl reports grants or contracts from the German Registry of COVID-19 Autopsies, funded by the Federal Ministry of Health and the Federal Ministry of Education and Research within the framework of the network of university medicine. Luiz Fernando Ferraz da Silva reports grants or contracts, paid to their institution, from the Bill and Melinda Gates Foundation. Martin Lammens reports support for the present manuscript from the Belgian Fund for Scientific Research–Flanders. Michael Osborn reports grants or contracts from the North West London pathology research grant (£10,000), paid by their own institution, to set up a tissue bank and fund the procurement and use of tissue included in the current research, as well as payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Yale University ($300 for talking about COVID). He also reports a leadership or fiduciary role on a board, society, committee, or advocacy group as president of the Royal College of Pathologists, secretary of the BDIAP, president AAPT (all unpaid). Paulo Hilario Nascimento Saldiva reports support for the present manuscript from the Bill and Melinda Gates Foundation, Conselho Nacional de Desenvolvimento Científico e Tecnológico, Fundação de Amparo à Pesquisa do Estado de São Paulo, and Hospital das Clinicas da Faculdade de Medicina da Universidade de Paulo–HC Convida. Tadaka Suzuki reports grants or contracts from the Japan Agency for Medical Research and Development and Japan Society for the Promotion of Science (grants in aid). There was no funding source for this study.
The Cardiac Autopsy in COVID-19 Study Group consists of Alberto E. Paniz Mondolfi (Department of Pathology, Molecular and Cell-Based Medicine, New York, New York), Aloke V. Finn (CVPath Institute, Inc., Gaithersburg, Maryland; and University of Maryland, Baltimore, Maryland), Amaro Nunes Duarte-Neto (Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil), Amy V. Rapkiewicz (NYU Winthrop Hospital, Department of Pathology, Long Island School of Medicine, Long Island, New York), Andrea Frustaci (Department of Clinical, Internal, Anesthesiologist and Cardiovascular Sciences, La Sapienza University, Rome, Italy; and Cellular and Molecular Cardiology Lab, IRCCS L. Spallanzani, Rome, Italy), Arthur-Atilla Keresztesi (Fogolyan Kristof Emergency County Hospital, Covasna County Institution of Forensic Medicine, Covasna, Romania), Brian Hanley (Department of Cellular Pathology, Northwest London Pathology, Imperial College London NHS Trust, London, UK; and Centre for Inflammatory Disease, Imperial College London, London, UK), Bruno Märkl (Institute of Pathology and Molecular Diagnostics, University Medical Center Augsburg, Augsburg, Germany), Christelle Lardi (University Center of Legal Medicine, Geneva University Hospital, Geneva, Switzerland), Clare Bryce (Icahn School of Medicine at Mount Sinai, New York, New York), Diana Lindner (Department of Cardiology, University Heart and Vascular Centre, Hamburg, Germany; and DZHK–German Center for Cardiovascular Research, Partner site, Hamburg/Kiel/Lübeck, Germany), Diego Aguiar (University Center of Legal Medicine, Geneva University Hospital, Geneva, Switzerland), Dirk Westermann (Department of Cardiology, University Heart and Vascular Centre, Hamburg, Germany; and DZHK–German Center for Cardiovascular Research, Partner site, Hamburg/Kiel/Lübeck, Germany), Edana Stroberg (Office of the Chief Medical Examiner, Oklahoma City, Oklahoma), Eric J. Duval (Office of the Chief Medical Examiner, Oklahoma City, Oklahoma), Esther Youd (Forensic Medicine and Science, University of Glasgow, Glasgow, UK), Gaetano Pietro Bulfamante (Unità di Anatomia Patologica, Dipartimento di Scienze della Salute, Università degli Studi di Milano, Milan, Italy; and Struttura Complessa di Anatomia Patologica e Genetica Medica, ASST Santi Paolo e Carlo, Milan, Italy), Isabelle Salmon (Department of Pathology, Erasme Hospital, Université Libre de Bruxelles (ULB), Brussels, Belgium; Centre Universitaire inter Régional d’expertise en Anatomie Pathologique Hospitalière, Jumet, Belgium; and DIAPath, Center for Microscopy and Molecular Imaging, ULB, Gosselies, Belgium), Johann Auer (Department of Cardiology and Intensive Care, St. Josef Hospital Braunau, Austria; and Department of Cardiology and Intensive Care, Kepler University of Medicine Linz, Austria), Joseph J. Maleszewski (Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota; and Department of Cardiovascular Medicine, Mayo Clinic, Rochester, Minnesota), Klaus Hirschbühl (Department of Hematology and Clinical Oncology, University Medical Center Augsburg, Augsburg, Germany), Lara Absil (Department of Pathology, Erasme Hospital, ULB, Brussels, Belgium), Lisa M. Barton (Office of the Chief Medical Examiner, Oklahoma City, Oklahoma), Luiz Fernando Ferraz da Silva (Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil; and Serviço de Verificação de Óbitos da Capital, Universidade de São Paulo, São Paulo, Brazil), Luiza Moore (Department of Histopathology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK; and Cancer, Ageing and Somatic Mutation, Wellcome Sanger Institute, Cambridge, UK), Marisa Dolhnikoff (Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil), Martin Lammens (Department of Pathology, Antwerp University Hospital, University of Antwerp, Edegem, Belgium), Melanie C. Bois (Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota), Michael Osborn (Department of Cellular Pathology, Northwest London Pathology, Imperial College London NHS Trust, London, UK; Death Investigation Committee, Royal College of Pathologists, London, UK; and Nightingale NHS Hospital, London, UK), Myriam Remmelink (Department of Pathology, Erasme Hospital, ULB, Brussels, Belgium), Paulo Hilario Nascimento Saldiva (Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil), Philippe G. Jorens (Infla-Med Research Consortium of Excellence, University of Antwerp, Antwerp, Belgium; Department of Medicine and Health Sciences, Laboratory of Experimental Medicine and Pediatrics, University of Antwerp, Antwerp, Belgium; and Department of Intensive Care Medicine, Antwerp University Hospital, University of Antwerp, Edegem, Belgium), Randall Craver (Children’s Hospital of New Orleans, New Orleans, Louisiana; and Louisiana State University Health Sciences Center, New Orleans, Louisiana), Renata Aparecida de Almeida Monteiro (Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil), Roberto Scendoni (Institute of Legal Medicine, Department of Law, University of Macerata, Macerata, Italy), Sanjay Mukhopadhyay (Department of Pathology, Cleveland Clinic, Cleveland, Ohio), Tadaki Suzuki (National Institute of Infectious Diseases, Tokyo, Japan), Thais Mauad (Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil), Tony Fracasso (University Center of Legal Medicine, Geneva University Hospital, Geneva, Switzerland), and Zachary Grimes (Icahn School of Medicine at Mount Sinai, New York, New York).
1. Mehta N., Kalra A., Nowacki A.S., Anjewierden S., Han Z., Bhat P., et al. Association of use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with testing positive for coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5:1020–1026. [PMC free article] [PubMed] [Google Scholar]
2. Reynolds H.R., Adhikari S., Pulgarin C., Troxel A.B., Iturrate E., Johnson S.B., et al. Renin-angiotensin-aldosterone system inhibitors and risk of COVID-19. N Engl J Med. 2020;382:2441–2448. [PMC free article] [PubMed] [Google Scholar]
3. Mancia G., Rea F., Ludergnani M., Apolone G., Corrao G. Renin-angiotensin-aldosterone system blockers and the risk of COVID-19. N Engl J Med. 2020;382:2431–2440. [PMC free article] [PubMed] [Google Scholar]
4. The Novel Coronavirus Pneumonia Emergency Response Epidemiology Team The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19)—China, 2020. China CDC Weekly. 2020;2:113–122. [PMC free article] [PubMed] [Google Scholar]
5. Xie Y., Xu E., Bowe B., Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. 2022;28:583–590. [PMC free article] [PubMed] [Google Scholar]
6. Dweck M.R., Bularga A., Hahn R.T., Bing R., Lee K.K., Chapman A.R., et al. Global evaluation of echocardiography in patients with COVID-19. Eur Heart J Cardiovasc Imaging. 2020;21:949–958. [PMC free article] [PubMed] [Google Scholar]
7. Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. [PMC free article] [PubMed] [Google Scholar]
8. Guo T., Fan Y., Chen M., Wu X., Zhang L., He T., et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5:811–818. [PMC free article] [PubMed] [Google Scholar]
9. Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. [PMC free article] [PubMed] [Google Scholar]
10. Shi S., Qin M., Shen B., Cai Y., Liu T., Yang F., et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020;5:802–810. [PMC free article] [PubMed] [Google Scholar]
11. Santoso A., Pranata R., Wibowo A., Al-Farabi M.J., Huang I., Antariksa B. Cardiac injury is associated with mortality and critically ill pneumonia in COVID-19: a meta-analysis. Am J Emerg Med. 2021;44:352–357. [PMC free article] [PubMed] [Google Scholar]
12. Kang Y., Chen T., Mui D., Ferrari V., Jagasia D., Scherrer-Crosbie M., et al. Cardiovascular manifestations and treatment considerations in COVID-19. Heart. 2020;106:1132. [PMC free article] [PubMed] [Google Scholar]
13. Libby P. The heart in COVID-19: primary target or secondary bystander? JACC Basic Transl Sci. 2020;5:537–542. [PMC free article] [PubMed] [Google Scholar]
14. Stefanini G.G., Montorfano M., Trabattoni D., Andreini D., Ferrante G., Ancona M., et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation. 2020;141:2113–2116. [PMC free article] [PubMed] [Google Scholar]
15. Chen C., Zhou Y., Wang D.W. SARS-CoV-2: a potential novel etiology of fulminant myocarditis. Herz. 2020;45:230–232. [PMC free article] [PubMed] [Google Scholar]
16. Inciardi R.M., Lupi L., Zaccone G., Italia L., Raffo M., Tomasoni D., et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5:819–824. [PMC free article] [PubMed] [Google Scholar]
17. Tian S., Xiong Y., Liu H., Niu L., Guo J., Liao M., et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. 2020;33:1007–1014. [PMC free article] [PubMed] [Google Scholar]
18. Lindner D., Fitzek A., Bräuninger H., Aleshcheva G., Edler C., Meissner K., et al. Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol. 2020;5:1281–1285. [PMC free article] [PubMed] [Google Scholar]
19. Wichmann D., Sperhake J.-P., Lütgehetmann M., Steurer S., Edler C., Heinemann A., et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. 2020;173:268–277. [PMC free article] [PubMed] [Google Scholar]
20. Grosse C., Grosse A., Salzer H.J.F., Dünser M.W., Motz R., Langer R. Analysis of cardiopulmonary findings in COVID-19 fatalities: high incidence of pulmonary artery thrombi and acute suppurative bronchopneumonia. Cardiovasc Pathol. 2020;49:107263. [PMC free article] [PubMed] [Google Scholar]
21. Edler C., Schröder A.S., Aepfelbacher M., Fitzek A., Heinemann A., Heinrich F., et al. Dying with SARS-CoV-2 infection-an autopsy study of the first consecutive 80 cases in Hamburg, Germany. Int J Leg Med. 2020;134:1275–1284. [PMC free article] [PubMed] [Google Scholar]
22. Duarte-Neto A.N., Monteiro R.A.A., da Silva L.F.F., Malheiros D., de Oliveira E.P., Theodoro-Filho J., et al. Pulmonary and systemic involvement in COVID-19 patients assessed with ultrasound-guided minimally invasive autopsy. Histopathology. 2020;77:186–197. [PMC free article] [PubMed] [Google Scholar]
23. Bois M.C., Boire N.A., Layman A.J., Aubry M.-C., Alexander M.P., Roden A.C., et al. COVID-19-associated nonocclusive fibrin microthrombi in the heart. Circulation. 2021;143:230–243. [PMC free article] [PubMed] [Google Scholar]
24. Bösmüller H., Traxler S., Bitzer M., Häberle H., Raiser W., Nann D., et al. The evolution of pulmonary pathology in fatal COVID-19 disease: an autopsy study with clinical correlation. Virchows Archiv. 2020;477:349–357. [PMC free article] [PubMed] [Google Scholar]
25. Bradley B.T., Maioli H., Johnston R., Chaudhry I., Fink S.L., Xu H., et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet. 2020;396:320–332. [PMC free article] [PubMed] [Google Scholar]
26. Fox S.E., Li G., Akmatbekov A., Harbert J.L., Lameira F.S., Brown J.Q., et al. Unexpected features of cardiac pathology in COVID-19 infection. Circulation. 2020;142:1123–1125. [PubMed] [Google Scholar]
27. Brook O.R., Piper K.G., Mercado N.B., Gebre M.S., Barouch D.H., Busman-Sahay K., et al. Feasibility and safety of ultrasound-guided minimally invasive autopsy in COVID-19 patients. Abdom Radiol (NY) 2021;46:1263–1271. [PMC free article] [PubMed] [Google Scholar]
28. Hanley B., Naresh K.N., Roufosse C., Nicholson A.G., Weir J., Cooke G.S., et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: a post-mortem study. Lancet Microbe. 2020;1:e245–e253. [PMC free article] [PubMed] [Google Scholar]
29. Jacobs W., Lammens M., Kerckhofs A., Voets E., Van San E., Van Coillie S., et al. Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail. 2020;7:3772–3781. [PMC free article] [PubMed] [Google Scholar]
30. Schurink B., Roos E., Radonic T., Barbe E., Bouman C.S.C., de Boer H.H., et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. 2020;1:e290–e299. [PMC free article] [PubMed] [Google Scholar]
31. Menter T., Haslbauer J.D., Nienhold R., Savic S., Hopfer H., Deigendesch N., et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;77:198–209. [PMC free article] [PubMed] [Google Scholar]
32. Remmelink M., De Mendonça R., D’Haene N., De Clercq S., Verocq C., Lebrun L., et al. Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit Care. 2020;24:495. [PMC free article] [PubMed] [Google Scholar]
33. Bulfamante G.P., Perrucci G.L., Falleni M., Sommariva E., Tosi D., Martinelli C., et al. Evidence of SARS-CoV-2 transcriptional activity in cardiomyocytes of COVID-19 patients without clinical signs of cardiac involvement. Biomedicines. 2020;8:626. [PMC free article] [PubMed] [Google Scholar]
34. Dolhnikoff M., Ferreira Ferranti J., de Almeida Monteiro R.A., Duarte-Neto A.N., Soares Gomes-Gouvêa M., Viu Degaspare N., et al. SARS-CoV-2 in cardiac tissue of a child with COVID-19-related multisystem inflammatory syndrome. Lancet Child Adolesc Health. 2020;4:790–794. [PMC free article] [PubMed] [Google Scholar]
35. Recalde-Zamacona B., García-Tobar L., Argueta A., Álvarez L., De Andrea C.E., Fernández Alonso M., et al. Histopathological findings in fatal COVID-19 severe acute respiratory syndrome: preliminary experience from a series of 10 Spanish patients. Thorax. 2020;75:1116–1118. [PMC free article] [PubMed] [Google Scholar]
36. Gauchotte G., Venard V., Segondy M., Cadoz C., Esposito-Fava A., Barraud D., et al. SARS-Cov-2 fulminant myocarditis: an autopsy and histopathological case study. Int J Leg Med. 2021;135:577–581. [PMC free article] [PubMed] [Google Scholar]
37. Martines R.B., Ritter J.M., Matkovic E., Gary J., Bollweg B.C., Bullock H., et al. Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg Infect Dis. 2020;26:2005–2015. [PMC free article] [PubMed] [Google Scholar]
38. Rapkiewicz A.V., Mai X., Carsons S.E., Pittaluga S., Kleiner D.E., Berger J.S., et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020;24:100434. [PMC free article] [PubMed] [Google Scholar]
39. Schaller T., Hirschbühl K., Burkhardt K., Braun G., Trepel M., Märkl B., et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323:2518–2520. [PMC free article] [PubMed] [Google Scholar]
40. Beigmohammadi M.T., Jahanbin B., Safaei M., Amoozadeh L., Khoshavi M., Mehrtash V., et al. Pathological findings of postmortem biopsies from lung, heart, and liver of 7 deceased COVID-19 patients. Int J Surg Pathol. 2021;29:135–145. [PMC free article] [PubMed] [Google Scholar]
41. del Nonno F., Frustaci A., Verardo R., Chimenti C., Nicastri E., Antinori A., et al. Virus-negative myopericarditis in human coronavirus infection. Circ Heart Fail. 2020;13 [PMC free article] [PubMed] [Google Scholar]
42. De Michele S., Sun Y., Yilmaz M.M., Katsyv I., Salvatore M., Dzierba A.L., et al. Forty postmortem examinations in COVID-19 patients. Am J Clin Pathol. 2020;154:748–760. [PMC free article] [PubMed] [Google Scholar]
43. Falasca L., Nardacci R., Colombo D., Lalle E., Di Caro A., Nicastri E., et al. Postmortem findings in Italian patients with COVID-19: a descriptive full autopsy study of cases with and without comorbidities. J Infect Dis. 2020;222:1807–1815. [PMC free article] [PubMed] [Google Scholar]
44. Basso C., Leone O., Rizzo S., De Gaspari M., van der Wal A.C., Aubry M.C., et al. Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J. 2020;41:3827–3835. [PMC free article] [PubMed] [Google Scholar]
45. Lax S.F., Skok K., Zechner P., Kessler H.H., Kaufmann N., Koelblinger C., et al. Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med. 2020;173:350–361. [PMC free article] [PubMed] [Google Scholar]
46. Guagliumi G., Sonzogni A., Pescetelli I., Pellegrini D., Finn A.V. Microthrombi and ST-segment–elevation myocardial infarction in COVID-19. Circulation. 2020;142:804–809. [PMC free article] [PubMed] [Google Scholar]
47. Buja L.M., Wolf D.A., Zhao B., Akkanti B., McDonald M., Lelenwa L., et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020;48:107233. [PMC free article] [PubMed] [Google Scholar]
48. Craver R., Huber S., Sandomirsky M., McKenna D., Schieffelin J., Finger L. Fatal eosinophilic myocarditis in a healthy 17-year-old male with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2c) Fetal Pediatr Pathol. 2020;39:263–268. [PMC free article] [PubMed] [Google Scholar]
49. Keresztesi A.A., Perde F., Ghita-Nanu A., Radu C.C., Negrea M., Keresztesi G. Post-mortem diagnosis and autopsy findings in SARS-CoV-2 infection: Forensic case series. Diagnostics. 2020;10:1070. [PMC free article] [PubMed] [Google Scholar]
50. Wang X.X., Shao C., Huang X.J., Sun L., Meng L.J., Liu H., et al. Histopathological features of multiorgan percutaneous tissue core biopsy in patients with COVID-19. J Clin Pathol. 2021;74:522. [PMC free article] [PubMed] [Google Scholar]
51. Youd E., Moore L. COVID-19 autopsy in people who died in community settings: the first series. J Clin Pathol. 2020;73:840. [PubMed] [Google Scholar]
52. Tanskanen M., Peuralinna T., Polvikoski T., Notkola I.L., Sulkava R., Hardy J., et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40:232–239. [PubMed] [Google Scholar]
53. González-López E., Gallego-Delgado M., Guzzo-Merello G., de Haro-del Moral F.J., Cobo-Marcos M., Robles C., et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–2594. [PubMed] [Google Scholar]
54. Bennani Smires Y., Victor G., Ribes D., Berry M., Cognet T., Méjean S., et al. Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: the high prevalence of amyloid cardiomyopathy. Int J Cardiovasc Imaging. 2016;32:1403–1413. [PubMed] [Google Scholar]
55. Mohammed S.F., Mirzoyev S.A., Edwards W.D., Dogan A., Grogan D.R., Dunlay S.M., et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2:113–122. [PMC free article] [PubMed] [Google Scholar]
56. Brannagan T.H., 3rd, Auer-Grumbach M., Berk J.L., Briani C., Bril V., Coelho T., et al. ATTR amyloidosis during the COVID-19 pandemic: insights from a global medical roundtable. Orphanet J Rare Dis. 2021;16:204. [PMC free article] [PubMed] [Google Scholar]
57. Pellegrini D., Kawakami R., Guagliumi G., Sakamoto A., Kawai K., Gianatti A., et al. Microthrombi as a major cause of cardiac injury in COVID-19. Circulation. 2021;143:1031–1042. [PubMed] [Google Scholar]
58. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. e8. [PMC free article] [PubMed] [Google Scholar]
59. Liu H., Gai S., Wang X., Zeng J., Sun C., Zhao Y., et al. Single-cell analysis of SARS-CoV-2 receptor ACE2 and spike protein priming expression of proteases in the human heart. Cardiovasc Res. 2020;116:1733–1741. [PMC free article] [PubMed] [Google Scholar]
60. Dong M., Zhang J., Ma X., Tan J., Chen L., Liu S., et al. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed Pharmacother. 2020;131:110678. [PMC free article] [PubMed] [Google Scholar]
61. Oudit G.Y., Kassiri Z., Jiang C., Liu P.P., Poutanen S.M., Penninger J.M., et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39:618–625. [PMC free article] [PubMed] [Google Scholar]
62. Pérez-Bermejo J.A., Kang S., Rockwood S.J., Simoneau C.R., Joy D.A., Ramadoss G.N., et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells predicts novel cytopathic features in hearts of COVID-19 patients. bioRxiv. 2020 [Epub ahead of print] [PMC free article] [PubMed] [Google Scholar]
63. Wong D.W.L., Klinkhammer B.M., Djudjaj S., Villwock S., Timm M.C., Buhl E.M., et al. Multisystemic cellular tropism of SARS-CoV-2 in autopsies of COVID-19 patients. Cells. 2021;10:1900. [PMC free article] [PubMed] [Google Scholar]
64. Auer J., Neuhierl F., Hetzmann Z. COVID-19-related fatal myocarditis in a 42-year-old female patient. Cardiol J. 2020;27:642–643. [PMC free article] [PubMed] [Google Scholar]
65. Bearse M., Hung Y.P., Krauson A.J., Bonanno L., Boyraz B., Harris C.K., et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod Pathol. 2021;34:1345–1357. [PMC free article] [PubMed] [Google Scholar]
66. Mason J.W., O’Connell J.B., Herskowitz A., Rose N.R., McManus B.M., Billingham M.E., et al. A clinical trial of immunosuppressive therapy for myocarditis. N Engl J Med. 1995;333:269–275. [PubMed] [Google Scholar]
67. Shanes J.G., Ghali J., Billingham M.E., Ferrans V.J., Fenoglio J.J., Edwards W.D., et al. Interobserver variability in the pathologic interpretation of endomyocardial biopsy results. Circulation. 1987;75:401–405. [PubMed] [Google Scholar]
68. Hauck A.J., Kearney D.L., Edwards W.D. Evaluation of postmortem endomyocardial biopsy specimens from 38 patients with lymphocytic myocarditis: implications for role of sampling error. Mayo Clin Proc. 1989;64:1235–1245. [PubMed] [Google Scholar]
69. Chow L.H., Radio S.J., Sears T.D., McManus B.M. Insensitivity of right ventricular endomyocardial biopsy in the diagnosis of myocarditis. J Am Coll Cardiol. 1989;14:915–920. [PubMed] [Google Scholar]
70. Katzmann J.L., Schlattmann P., Rigopoulos A.G., Noutsias E., Bigalke B., Pauschinger M., et al. Meta-analysis on the immunohistological detection of inflammatory cardiomyopathy in endomyocardial biopsies. Heart Fail Rev. 2020;25:277–294. [PubMed] [Google Scholar]
71. Halushka M.K., Vander Heide R.S. Myocarditis is rare in COVID-19 autopsies: cardiovascular findings across 277 postmortem examinations. Cardiovasc Pathol. 2021;50:107300. [PMC free article] [PubMed] [Google Scholar]
72. Wojnicz R., Nowalany-Kozielska E., Wojciechowska C., Glanowska G., Wilczewski P., Niklewski T., et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy. Circulation. 2001;104:39–45. [PubMed] [Google Scholar]
73. Ojha V., Verma M., Pandey N.N., Mani A., Malhi A.S., Kumar S., et al. Cardiac magnetic resonance imaging in coronavirus disease 2019 (COVID-19): a systematic review of cardiac magnetic resonance imaging findings in 199 patients. J Thorac Imaging. 2021;36:73–83. [PubMed] [Google Scholar]
74. Basso C., Aguilera B., Banner J., Cohle S., d’Amati G., de Gouveia R.H., et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017;471:691–705. [PMC free article] [PubMed] [Google Scholar]
75. Caforio A.L.P., Pankuweit S., Arbustini E., Basso C., Gimeno-Blanes J., Felix S.B., et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34:2636–2648. [PubMed] [Google Scholar]
Big Pharma Lobbied Social Media to Flag COVID ‘Misinformation,’ Latest ‘Twitter Files’ Reveal
COVID-19 vaccine manufacturers, including Pfizer, BioNTech and Moderna, lobbied Twitter and other social media platforms to set moderation rules that would flag purported COVID-19-related “misinformation,” according to Lee Fang, who reported on the latest “Twitter files.” By Michael Nevradakis, Ph.D. 9
Link copied twitter files big pharma covid feature
Miss a day, miss a lot. Subscribe to The Defender’s Top News of the Day. It’s free.
COVID-19 vaccine manufacturers, including Pfizer, BioNTech and Moderna, lobbied Twitter and other social media platforms to set moderation rules that would flag purported COVID-19-related “misinformation,” according to the latest installment of the “Twitter files.”
Published Jan. 16 on Twitter and in The Intercept by investigative journalist Lee Fang, the documents revealed efforts by lobbying groups, nonprofits and the German government to influence Twitter’s content moderation policies to more aggressively limit purported COVID-19 “misinformation.”
The lobbying efforts also focused on limiting social media posts that urged pharmaceutical companies to renounce their intellectual property rights over COVID-19 vaccines and therapeutics to facilitate the development of low-cost generic equivalents, which would potentially have cost these companies billions of dollars in profits.
In releasing this installment of the “Twitter files,” Fang noted that he “was given some access to Twitter emails” and that the searches for these documents “were carried out by a Twitter attorney,” potentially limiting the information made available to him.
Big Pharma ‘lobbied social media to shape content around vaccine policy’
According to Fang, “the pharmaceutical industry lobbied social media to shape content around vaccine policy.”
One example came via BIO (Biotechnology Innovation Organization), described by Fang as “Pfizer & Moderna’s lobbying group.” Fang wrote that BIO “fully funded a special content moderation campaign designed by a contractor called Public Good Projects [PGP], which worked [with] Twitter to set content moderation rules around COVID ‘misinformation.’”
Washington, D.C.-based BIO operates websites such as “COVID Vaccine Facts” and claims it promotes the “research and development of innovative healthcare, agricultural, industrial and environmental biotechnology products.”
PGP describes itself as “a public health nonprofit specializing in large-scale media monitoring programs, social and behavior change interventions, and cross-sector initiatives” that “is led by experts in public health, marketing, journalism, media, and business” who “create bold projects for health.”
PGP CEO Joe Smyser, Ph.D., previously completed a postdoctoral study at the Centers for Disease Control and Prevention and an internship at the U.S. Food and Drug Administration.
BUY NOW: Ed Dowd’s Must-Read Book — “Cause Unknown”
Other members of PGP’s board have previously held leadership positions for the U.S. Department of Health and Human Services, Discovery Channel, Levi Strauss & Co., Merck, Morgan Stanley, Pepsi-Cola, Sesame Workshop (producers of Sesame Street), The Advertising Council, TikTok, Tumblr, the U.S. Agency for International Development, the U.S. Army and the World Health Organization.
PGP’s “recent partners” include Google, The Rockefeller Foundation, UNICEF and the Yale Institute for Global Health. As previously reported by The Defender, The Rockefeller Foundation partnered with nonprofit organizations to fund behavioral psychology research intended to “nudge” more people to get COVID-19 vaccines.
According to tax forms published by Fang, BIO funded the “nudge” campaign, titled “Stronger,” to the tune of $1.275 million. The “Stronger” campaign “helped Twitter create content moderation bots, select which public health accounts got verification [and] helped crowdsource content takedowns,” wrote Fang on Twitter.
Fang wrote that PGP “regularly communicated with Twitter on regulating content related to the pandemic” and helped “develop bots to censor vaccine misinformation.” PGP also sometimes “sent direct requests to Twitter with lists of accounts to censor and verify.”
BIO’s funding also helped the “Stronger” campaign develop “tools for the public to flag content on Twitter, Instagram, and Facebook for moderation,” according to Fang.
Some of the “regular correspondence” between PGP and Twitter “throughout 2021 and 2022” was channeled to Todd O’Boyle, formerly Twitter’s senior manager of public policy. O’Boyle “served as a point of contact with the Biden administration,” Fang reported.
While Fang claimed that some of the tweets the “Stronger” campaign focused on were, in his words, “truly unhinged misinfo, like claims that vaccines include microchips,” many others “were more of a grey area, like vaccine passports [and] vaccine mandates” and “policies that coerce vaccination.”
Writing in The Intercept, Fang said that one such tweet that was flagged stated:
“If a vaccinated person and an unvaccinated person have roughly the same capacity to carry, shed and transmit the virus, particularly in its Delta form, what difference does implementing a vaccination passport actually make to the spread of the virus?”
According to Fang, this campaign sent “direct regular emails” to Twitter, “with lists of tweets to takedown [sic] & others to verify.” He added that many of these requests “focused on @zerohedge, which was suspended.”
A screenshot of a Feb. 24, 2022, email from PGP to O’Boyle and presumably others at Twitter contained “this week’s misinfo report” and “our batch verification request.”
Smyser, in remarks provided to Fang, described the “Stronger” campaign as a good-faith effort to combat disinformation, describing his job as “how do people figure out where to go get vaccinated? And how do I encourage them to get the vaccine?”
Fang noted that the “Stronger” campaign did not apply to the pharmaceutical companies, such as when they “wildly exaggerated the risks of creating low-cost generic COVID vaccines” — another focus of Fang’s “Twitter files” drop.
“The rules applied only to critics of industry,” Fang wrote.
Fang noted that lobbying efforts on the part of COVID-19 vaccine manufacturers against the production of low-cost generic equivalents afforded those companies massive revenues, as they “saw the crisis as an opportunity for unprecedented profit.”
According to Fang:
“The largely successful assault against the creation of generic vaccines resulted in an unprecedented explosion in profit for a few select biopharmaceutical drug interests. Pfizer and BioNTech generated a staggering $37 billion in revenue from its shared mRNA vaccine in 2021 alone, making it one of the most lucrative drug products of all time.
“Moderna, which made $17.7 billion from vaccine sales in 2021, recently announced its plan to hike the price of its COVID shot by about 400 percent.”
As part of these lobbying efforts, BIO reached out to the newly elected Biden administration to advocate in favor of U.S. sanctions against “any country attempting to violate patent rights and create generic low-cost COVID medicine or vaccines.”
BioNTech, a German pharmaceutical company that jointly developed a COVID-19 vaccine with Pfizer, “reached out to Twitter to request that Twitter directly censor users tweeting at them to ask for generic low cost vaccines,” wrote Fang, who added that this request “was also backed by the German government.”
Fang shared a screenshot of a Dec. 13, 2020, email from Nina Morschhaeuser, head of Public Policy, Government and Philanthropy for Twitter Germany, stating that she had been contacted by BioNTech with a warning about a forthcoming “campaign targeting the pharmaceutical companies developing the COVID-19 vaccine.”
BioNTech asked Twitter to “hide” such tweets over a period of two days, wrote Fang, and to monitor such accounts, including allegedly “fake accounts” that Fang revealed belonged to “real people,” including a “74-year-old retired bricklayer in the U.K.”
A website affiliated with the CDU/CSU, an alliance of Germany’s Christian Democratic Union and Christian Social Union, promoted Morschhaeuser’s speaking engagements. Former German Chancellor Angela Merkel, a member of the CDU, imposed some of Europe’s strictest COVID-19-related restrictions.
Release of ‘Fauci files’ still pending, as House investigation into federal interference launched
Although, in a Jan. 2 tweet, Twitter owner and CEO Elon Musk said the publication of the “Fauci files,” as part of the ongoing release of the “Twitter files,” would come “later this week,” they still have not been publicized as of the time of this writing.
Responding to Musk in a Jan. 8 appearance on CBS’ “The Takeout,” Dr. Anthony Fauci said, “I have no idea what [Musk]’s talking about. I mean, there’s a lot of misinformation, conspiracy theories and disinformation going on. I have nothing to say to him. I don’t understand what he’s doing. It’s just unfortunate.”
Other “Twitter files” releases in the past week involved further censorship requests by Rep. Adam Schiff (D-Calif.), including a request for the removal of a parody video of Joe Biden retweeted by then-President Donald Trump, and “Russiagate lies,” revealing pressure on Twitter from prominent Democratic Party officials to connect tweets under the “#ReleaseTheMemo” hashtag, to “Russian bots.”
On Jan. 10, the Republican-controlled House of Representatives voted 221-211 to create a new Select Committee on the Weaponization of the Federal Government,” responding in part to other previous “Twitter files” releases that revealed pressure on Twitter from federal agencies to censor or deplatform specific users on the platform.
Rep. Jim Jordan (R-Ohio), who will head the committee, on Jan. 8 told “Fox News Sunday”:
“We have a duty to get into these agencies and look at how they have been weaponized to go against the very people they’re supposed to represent, how they have infringed on First Amendment liberties of the American people. And we’re going to do that.
“We’re going to do it in a way that’s consistent with the Constitution. But we’re going to do it vigorously. We’re going to do it aggressively. Because that’s our job.”
Separately from the “Twitter files,” Revolver News revealed, on Jan. 6, details about a “mini-network” of fake “doctors” created early during the pandemic to “stoke fear over COVID-19” by claiming they lost loved ones due to COVID-19.
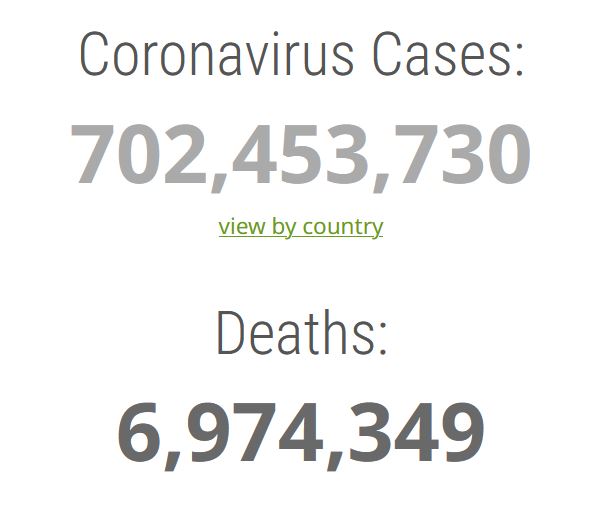
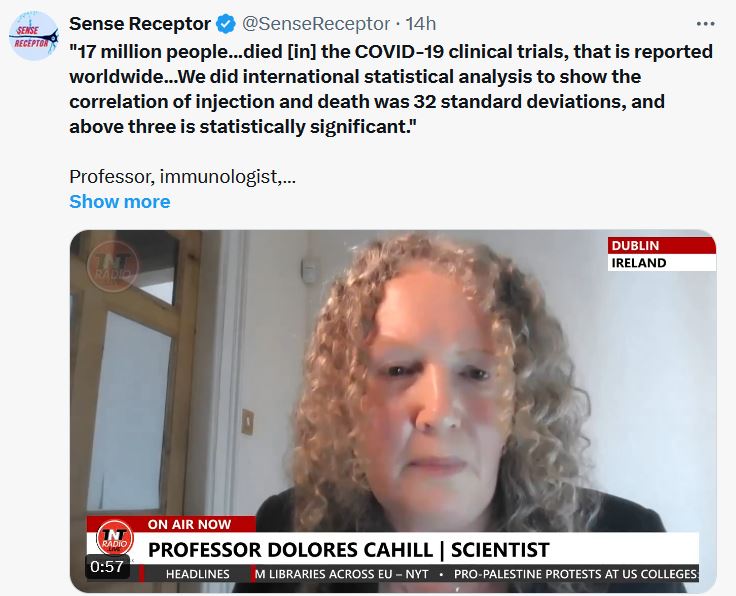







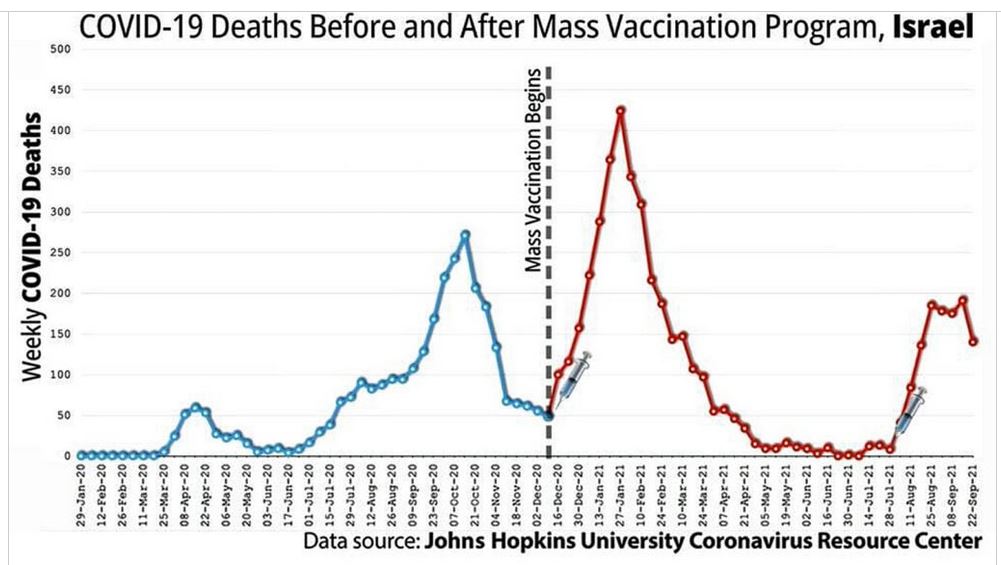
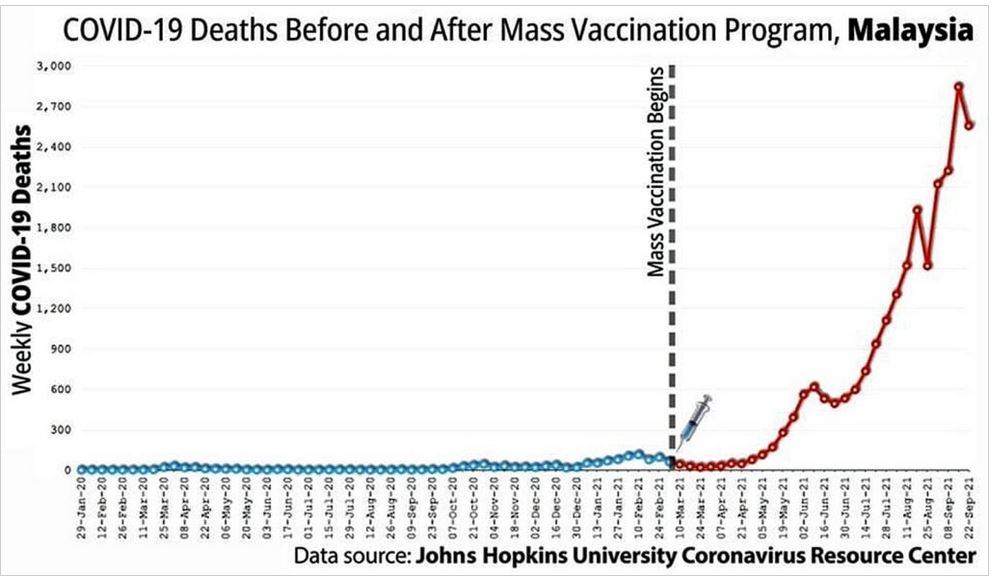

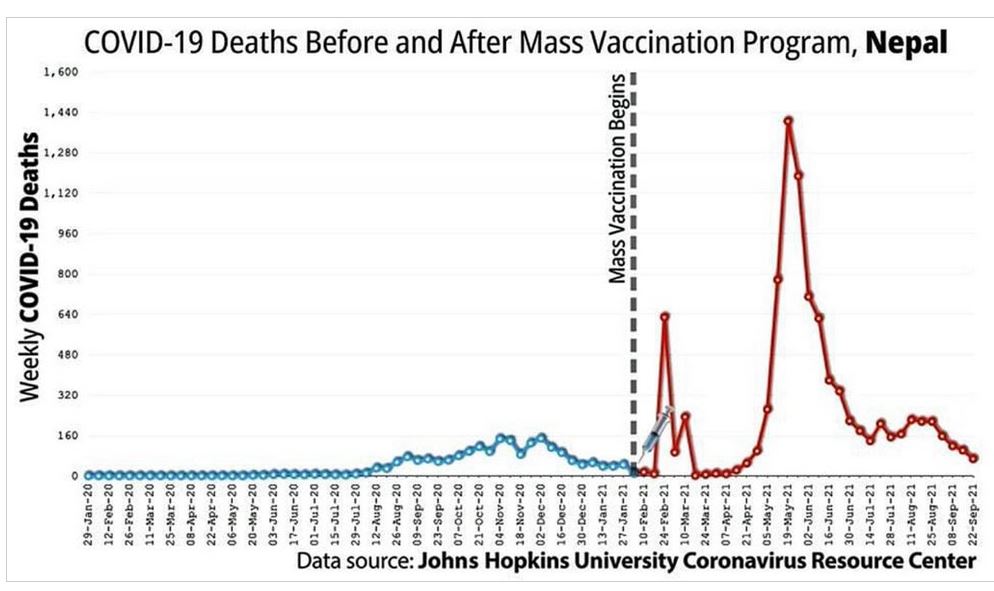
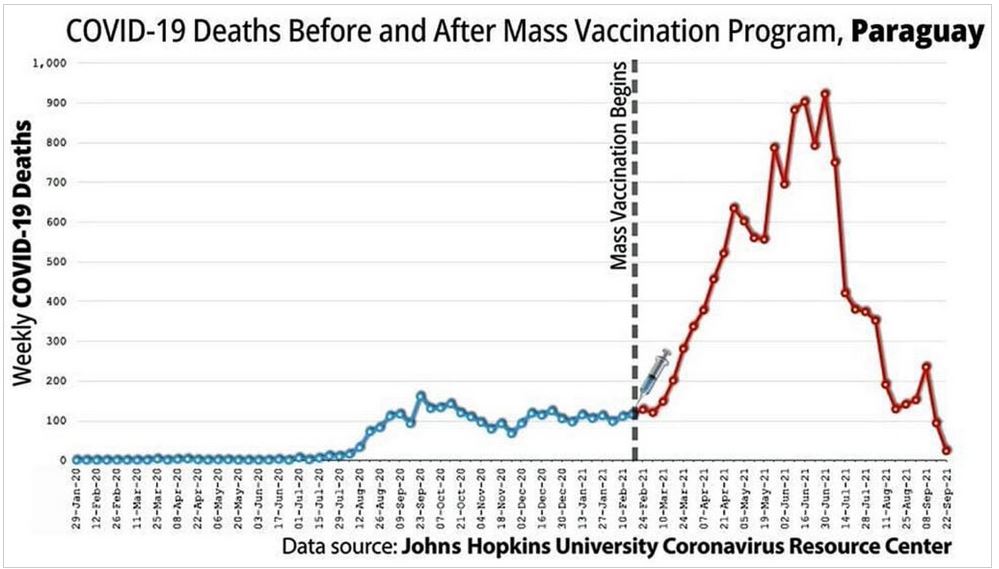
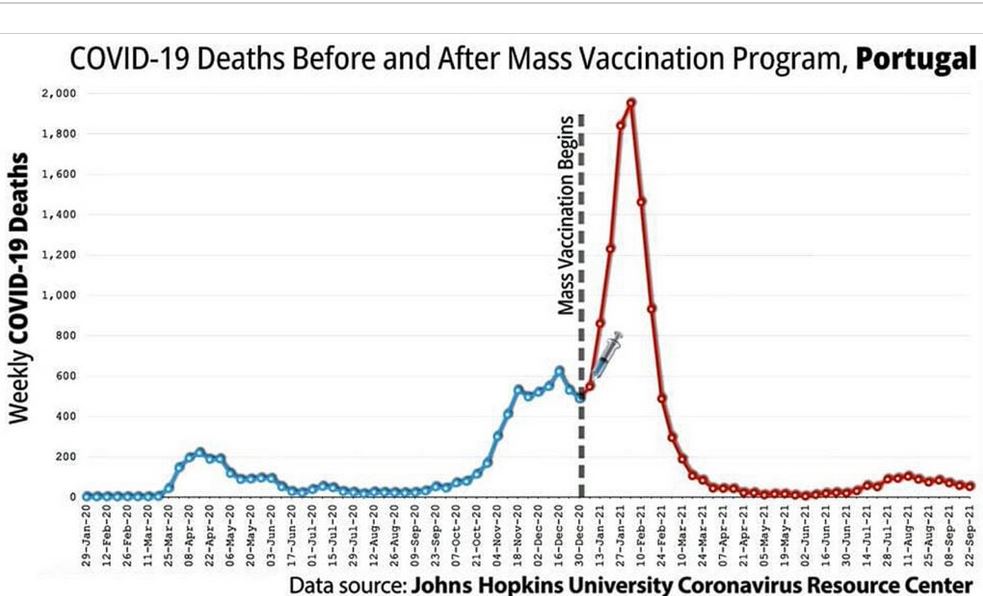
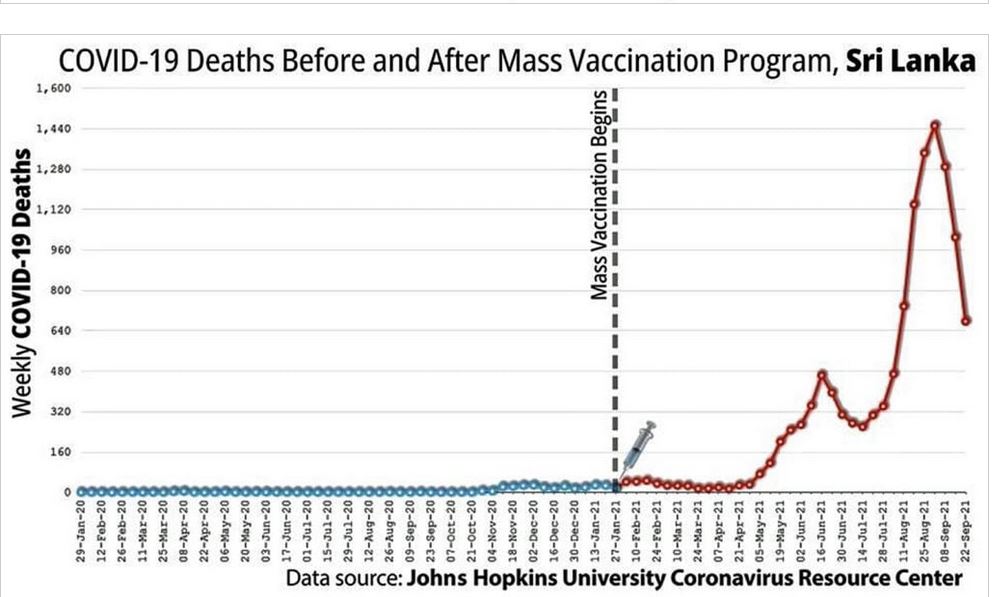
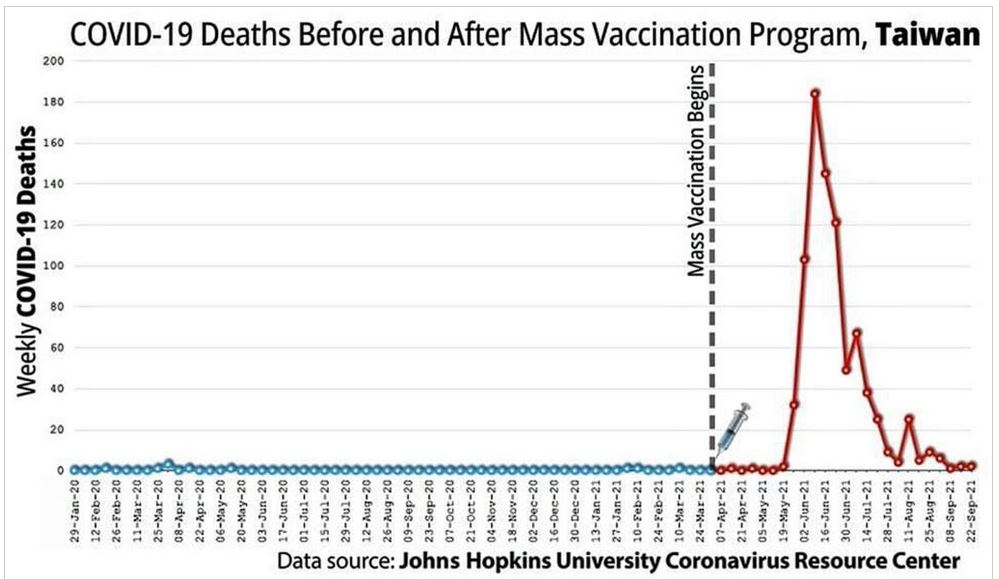
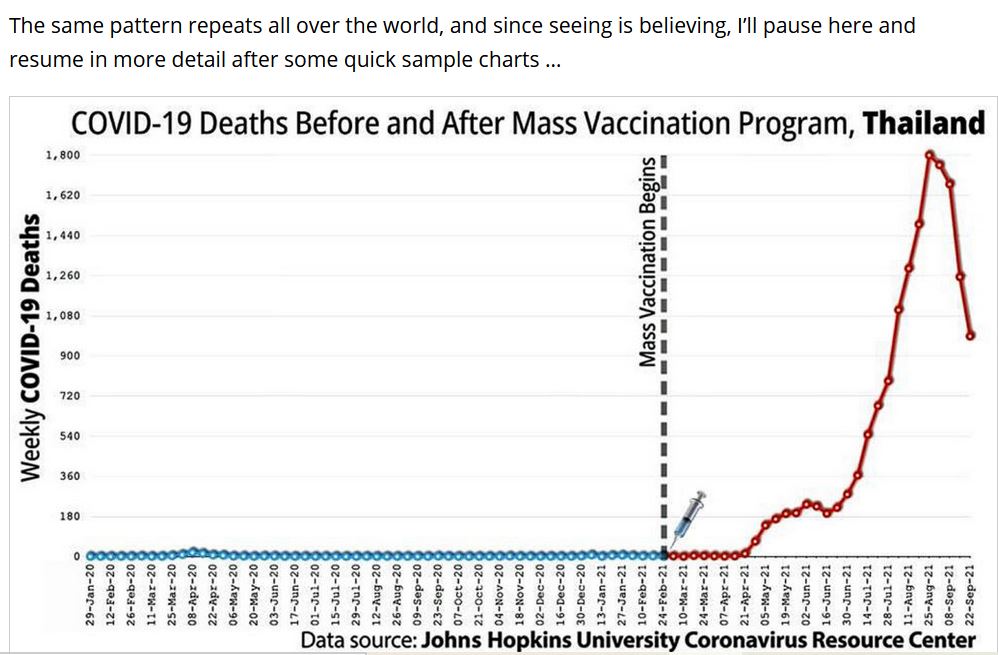
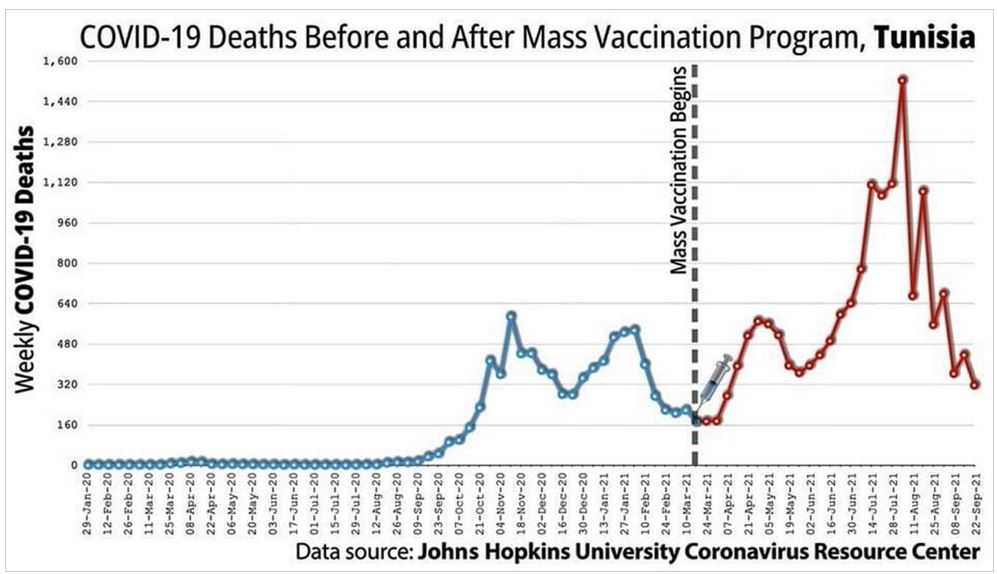
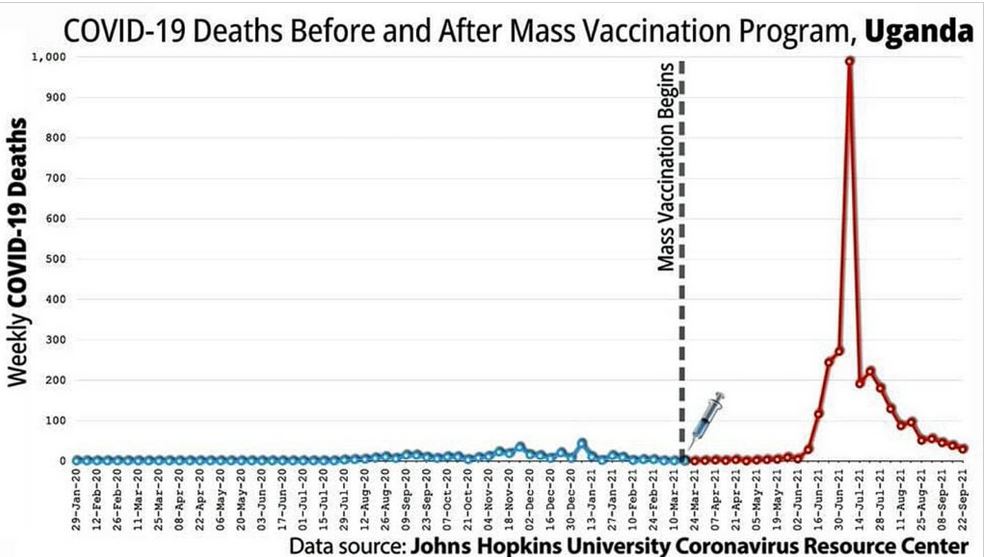
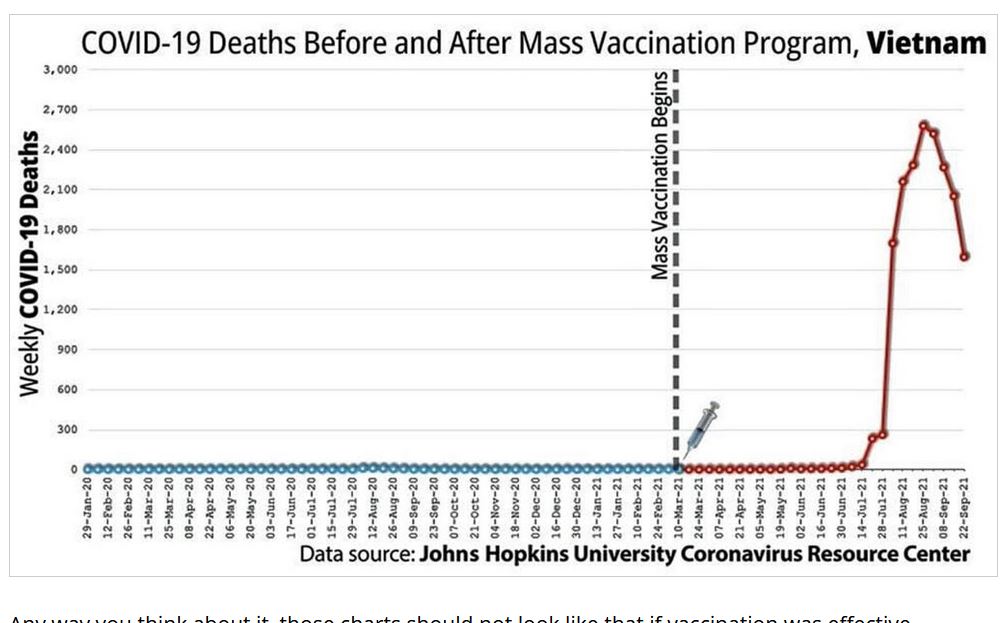

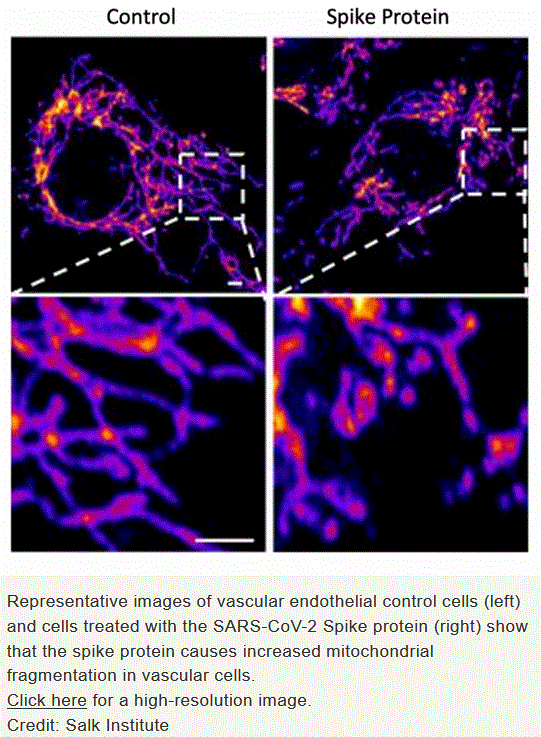


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}